Regulation of the Cardiovascular System
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
88 Terms
regulation of the cardiovascular system
Describe the roles of the medullary cardiovascular centers, hypothalamus, and cortex in the autonomic regulation of cardiac and vascular function.
Describe the origin and distribution of sympathetic and parasympathetic nerves to the heart and circulation.
Know the location and function of alpha- and beta-adrenoceptors and muscarinic receptors in the heart and blood vessels.
Describe the effects of sympathetic and parasympathetic stimulation on the heart and circulation.
List the anatomical components of the baroreceptor reflex.
Describe how carotid sinus baroreceptors respond to changes in arterial pressure (mean pressure and pulse pressure), and explain how changes in baroreceptor activity affect sympathetic and parasympathetic outflow to the heart and circulation.
Explain the sequence of events mediated by cardiopulmonary (volume) receptors that occur after an acute increase or decrease in arterial blood pressure.
Describe (a) the location of peripheral and central chemoreceptors; (b) the way they respond to hypoxemia, hypercapnia, and acidosis, and 9c) the effects of their stimulation in autonomic control of the heart and circulation.
List the factors that stimulate the release of catecholamines, renin, atrial natriuretic peptide, and vasopressin.
Describe how the sympathetic nerves, circulating catecholamines, angiotensin II, aldosterone, atrial natriuretic peptide, and vasopressin interact to regulate arterial blood pressure.
Describe the autonomic and hormonal compensatory mechanisms that are activated to restore arterial pressure following hemorrhage.
List the mechanisms and causes of hypotension.
Describe the causes of hypertension and its relationship with the Renin-Angiotensin Aldosterone System.
List the drugs used in the treatment of hypertension and their targets.
Describe the mechanism of neurohumoral activation in heart failure.
Describe the cardiovascular responses to exercise and acute stress (fight-or-flight response and vasovagal syncope).
Define autoregulation of blood flow, reactive hyperemia and active (functional) hyperemia.
Describe the blood flow regulation mechanisms in major vascular beds of the body such as: renal, cerebral, and coronary circulations.
constriction for baroreceptors leads means a decrease in pressure because there is less stretching.
however, VASOconstriction increases pressure.
Constrict Carotids (constriction= less pressure= less stretcing)→ ↓ Pressure in Carotid Sinus → ↓ Baroreceptor Firing → Medulla (NTS/VMC) → ↑ Sympathetic Outflow & ↓ Parasympathetic Outflow → ↑ Heart Rate & ↑ Vasoconstriction → ↑ Arterial Pressure
what is the cardiovascular system?
The cardiovascular system is a closed network composed of the heart, blood, and blood vessels. Its primary function is to deliver essential substances to cells and remove metabolic waste products.
Describe the roles of the medullary cardiovascular centers, hypothalamus, and cortex in the autonomic regulation of cardiac and vascular function. (learning objective)
medullary cardiovascular centers
hypothalamus
cortex
The autonomic regulation of cardiac and vascular function is a hierarchical system, with each level—Medullary Cardiovascular Centers, Hypothalamus, and Cortex—playing a distinct and integrated role.
Here is a description of their roles, from the most reflexive to the most complex.
1. Medullary Cardiovascular Centers: The Central Processing Unit (Both sympathetic and parasympathetic)
“medullary”: located in the medulla oblangata
Located in the medulla oblongata of the brainstem, this is the primary and most fundamental center for cardiovascular control. It integrates sensory input and sends coordinated autonomic output to the heart and blood vessels. The medullary cardiovascular center consists of two main components:
Cardio-inhibitory Center (or Nucleus Ambiguus):
“cardio-inhibitory center”= parasympethetic
Function: Primarily responsible for parasympathetic (vagal) output.
Action: Sends signals via the vagus nerve (CN X) to the heart, specifically the SA and AV nodes.
Effect: Decreases heart rate (negative chronotropy) and reduces the force of contraction (slightly) because it’s parasympathetic. This is the "brake" on the heart, promoting "rest-and-digest" functions.
Cardio-acceleratory Center & Vasomotor Center:
cardio-acceleratory center: sympathetic
Function: Primarily responsible for sympathetic output.
Action:
Cardiac: Sends signals to the spinal cord, which then relays them via sympathetic nerves to the heart, increasing heart rate (positive chronotropy) and contractility (positive inotropy).
Vascular: The Vasomotor Center specifically controls the tone (degree of constriction) of arterioles.
The Vasomotor Center sends a constant low level of sympathetic signal (vasomotor tone). Increasing the sympathetic signal causes vasoconstriction (for faster blood flow); decreasing the sympathetic system causes vasodilation.
Effect: The Cardio-acceleratory Center & Vasomotor Center is the The "accelerator," preparing the body for "fight-or-flight" by increasing cardiac output and blood pressure.
Key Input to the Medullary Centers: The Baroreceptor Reflex
The medulla is the central processor for this critical short-term BP regulation.
Sensors: Baroreceptors in the carotid sinuses and aortic arch detect changes in blood pressure.
Process: A drop in BP reduces baroreceptor firing.
Medullary Response: The cardio-inhibitory center is suppressed (less vagal activity), and the vasomotor center is activated (more sympathetic activity).
Result: Heart rate and contractility increase, and widespread vasoconstriction occurs, rapidly restoring blood pressure.
sympathetic= vasoconstriction to increase blood pressure
parasympathetic= vasodilation to decrease blood pressure
reduction in baroreceptor firing causes activation of sympathetic nervous system= vasoconstriction.
In summary, the Medullary Centers provide minute-to-minute, reflexive control of heart rate, contractility, and vascular tone.
2. Hypothalamus: The Integrative and Command Center
The hypothalamus sits above the brainstem (above the pituitary gland, NOT above the medulla oblongata) and the hypothalamus integrates 1. autonomic, 2. endocrine, and 3. behavioral responses. It does not replace the medullary centers but the hypothalamus commands and modulates them to serve broader physiological and emotional goals.
Autonomic Integration: The hypothalamus has direct neural connections to the medullary cardiovascular centers. the hypothalamus can override local reflexes to coordinate cardiovascular function with:
Temperature Regulation: In response to heat (too hot), the hypothalamus inhibits the vasomotor center, leading to cutaneous vasodilation to dissipate heat. In response to cold (too hot), it stimulates vasoconstriction to conserve heat.
Stress & "Fight-or-Flight" Response: During perceived threat or stress, the hypothalamus activates the sympathetic nervous system via the medullary centers and the spinal cord. This causes a massive increase in heart rate, contractility, and vasoconstriction in non-essential beds (e.g., gut), shunting blood to muscles.
vasoconstriction to NON-ESSENTIAL beds so more blood flows to muscles.
Defense Reaction: Specific hypothalamic areas, when stimulated, can evoke a pattern of cardiovascular changes (like increased BP and heart rate) associated with anger or fear.
Circadian Rhythm: The hypothalamus helps set the daily (circadian) rhythm of blood pressure, which is typically higher during the day and lower at night.
Endocrine Coordination: The hypothalamus regulates the pituitary gland, which in turn controls hormones that affect cardiovascular function (e.g., Vasopressin/ADH, which causes vasoconstriction and water retention).
vasopressin/ADH causes vasoCONSTRICTION and WATER RETENTION.
In summary, the Hypothalamus provides a higher level of control, linking cardiovascular function to temperature, emotion, stress, and overall homeostasis.
3. Cortex: The Executive Decision-Maker
The cerebral cortex, particularly the limbic system (emotion) and prefrontal cortex (planning, decision-making), provides the highest level of influence. It is responsible for psychosomatic and volitional influences on the cardiovascular system.
Emotional Influence (Limbic System):
Feelings of anxiety, excitement, or fear can trigger a cortical response that projects to the hypothalamus, leading to a rapid increase in heart rate and blood pressure (e.g., feeling your heart pound before a public speech).
Conversely, feelings of calm or meditation can activate parasympathetic pathways, lowering heart rate and blood pressure.
Volitional (Conscious) and Anticipatory Control:
The mere anticipation of exercise (the "get-ready" phase) can cause a cortical-mediated increase in heart rate and cardiac output via the hypothalamus and medullary centers.
Conditioned responses (e.g., a phobia of something that causes a cardiovascular reaction) are processed by the cortex and amygdala.
Cognitive Appraisal of Stress: How the cortex interprets a situation determines the magnitude of the cardiovascular response.
In summary, the Cortex links conscious thought, emotion, and learned behavior to cardiovascular function, allowing for anticipatory and psychologically-driven changes in heart rate and blood pressure.
Summary Table: Hierarchical Regulation
Level | Primary Role | Example of Action |
|---|---|---|
Cortex | Executive/Volitional | Heart rate rises due to anxiety before an exam; anticipatory rise before exercise. |
Hypothalamus | Integrative/Command | Redirects blood flow for thermoregulation; coordinates "fight-or-flight" response. |
Medullary Centers | Reflexive/Homeostatic | Baroreceptor reflex instantly increases heart rate when you stand up to prevent fainting. |
This hierarchical system ensures that the cardiovascular system can respond with precise reflexes for immediate survival (medulla), be integrated into broader physiological programs (hypothalamus), and be influenced by our thoughts, emotions, and environment (cortex).
1. Medullary Cardiovascular Centers: The Central Processing Unit (Both sympathetic and parasympathetic)
Cardio-acceleratory Center & Vasomotor Center (sympathetic)
Cardio-inhibitory Center (or Nucleus Ambiguus) (parasympathetic)
1. Medullary Cardiovascular Centers: The Central Processing Unit (Both sympathetic and parasympathetic)
“medullary”: located in the medulla oblangata
Located in the medulla oblongata of the brainstem, this is the primary and most fundamental center for cardiovascular control. It integrates sensory input and sends coordinated autonomic output to the heart and blood vessels. The medullary cardiovascular center consists of two main components:
Cardio-inhibitory Center (or Nucleus Ambiguus):
“cardio-inhibitory center”= parasympethetic
Function: Primarily responsible for parasympathetic (vagal) output.
Action: Sends signals via the vagus nerve (CN X) to the heart, specifically the SA and AV nodes.
Effect: Decreases heart rate (negative chronotropy) and reduces the force of contraction (slightly) because it’s parasympathetic. This is the "brake" on the heart, promoting "rest-and-digest" functions.
Cardio-acceleratory Center & Vasomotor Center:
cardio-acceleratory center: sympathetic
Function: Primarily responsible for sympathetic output.
Action:
Cardiac: Sends signals to the spinal cord, which then relays them via sympathetic nerves to the heart, increasing heart rate (positive chronotropy) and contractility (positive inotropy).
Vascular: The Vasomotor Center specifically controls the tone (degree of constriction) of arterioles.
The Vasomotor Center sends a constant low level of sympathetic signal (vasomotor tone). Increasing the sympathetic signal causes vasoconstriction (for faster blood flow); decreasing the sympathetic system causes vasodilation.
Effect: The Cardio-acceleratory Center & Vasomotor Center is the The "accelerator," preparing the body for "fight-or-flight" by increasing cardiac output and blood pressure.
Key Input to the Medullary Centers: The Baroreceptor Reflex
The medulla is the central processor for this critical short-term BP regulation.
Sensors: Baroreceptors in the carotid sinuses and aortic arch detect changes in blood pressure.
Process: A drop in BP reduces baroreceptor firing.
Medullary Response: The cardio-inhibitory center is suppressed (less vagal activity), and the vasomotor center is activated (more sympathetic activity).
Result: Heart rate and contractility increase, and widespread vasoconstriction occurs, rapidly restoring blood pressure.
sympathetic= vasoconstriction to increase blood pressure
parasympathetic= vasodilation to decrease blood pressure
reduction in baroreceptor firing causes activation of sympathetic nervous system= vasoconstriction.
In summary, the Medullary Centers provide minute-to-minute, reflexive control of heart rate, contractility, and vascular tone.
2. Hypothalamus: The Integrative and Command Center
relationship between hypothalamus and medullary centers
temperature regulation
stress and “fight or flight”
defense reaction
circadian rhythm
circadian rhythm
endocrine coordination
2. Hypothalamus: The Integrative and Command Center
The hypothalamus sits above the brainstem (above the pituitary gland, NOT above the medulla oblongata) and the hypothalamus integrates 1. autonomic, 2. endocrine, and 3. behavioral responses. It does not replace the medullary centers but the hypothalamus commands and modulates them to serve broader physiological and emotional goals.
Autonomic Integration: The hypothalamus has direct neural connections to the medullary cardiovascular centers. the hypothalamus can override local reflexes to coordinate cardiovascular function with:
Temperature Regulation: In response to heat (too hot), the hypothalamus inhibits the vasomotor center, leading to cutaneous vasodilation to dissipate heat. In response to cold (too hot), it stimulates vasoconstriction to conserve heat.
Stress & "Fight-or-Flight" Response: During perceived threat or stress, the hypothalamus activates the sympathetic nervous system via the medullary centers and the spinal cord. This causes a massive increase in heart rate, contractility, and vasoconstriction in non-essential beds (e.g., gut), shunting blood to muscles.
vasoconstriction to NON-ESSENTIAL beds so more blood flows to muscles.
Defense Reaction: Specific hypothalamic areas, when stimulated, can evoke a pattern of cardiovascular changes (like increased BP and heart rate) associated with anger or fear.
Circadian Rhythm: The hypothalamus helps set the daily (circadian) rhythm of blood pressure, which is typically higher during the day and lower at night.
Endocrine Coordination: The hypothalamus regulates the pituitary gland, which in turn controls hormones that affect cardiovascular function (e.g., Vasopressin/ADH, which causes vasoconstriction and water retention).
vasopressin/ADH causes vasoCONSTRICTION and WATER RETENTION.
In summary, the Hypothalamus provides a higher level of control, linking cardiovascular function to temperature, emotion, stress, and overall homeostasis.
3. Cortex: The Executive Decision-Maker
emotional influence (limbic system)
anticipation
3. Cortex: The Executive Decision-Maker
The cerebral cortex, particularly the limbic system (emotion) and prefrontal cortex (planning, decision-making), provides the highest level of influence. It is responsible for psychosomatic and volitional influences on the cardiovascular system.
limbic system: a complex system of nerves and networks in the brain, involving several areas near the edge of the cortex concerned with instinct and mood. It controls the basic emotions (fear, pleasure, anger) and drives (hunger, sex, dominance, care of offspring).
Emotional Influence (Limbic System):
Feelings of anxiety, excitement, or fear can trigger a cortical response that projects to the hypothalamus, leading to a rapid increase in heart rate and blood pressure (e.g., feeling your heart pound before a public speech).
Conversely, feelings of calm or meditation can activate parasympathetic pathways, lowering heart rate and blood pressure.
Volitional (Conscious) and Anticipatory Control:
The mere anticipation of exercise (the "get-ready" phase) can cause a cortical-mediated increase in heart rate and cardiac output via the hypothalamus and medullary centers.
Conditioned responses (e.g., a phobia of something that causes a cardiovascular reaction) are processed by the cortex and amygdala.
Cognitive Appraisal of Stress: How the cortex interprets a situation determines the magnitude of the cardiovascular response.
In summary, the Cortex links conscious thought, emotion, and learned behavior to cardiovascular function, allowing for anticipatory and psychologically-driven changes in heart rate and blood pressure.
functions of the vasomotor center (Medullary Cardiovascular Centers)
vasomotor tone
lateral portion of vmc: sympathetic
medial portion of vmc: parasympathetic
1. Control of Blood Vessels: Vasomotor Tone
maintaining the vasomotor tone is the fundamental job of the VMC. The vasoconstrictor area (lateral part) sends a constant, low-frequency stream of signals through the sympathetic nerves to the smooth muscle in blood vessel walls.
This baseline level of stimulation, known as vasomotor tone, keeps the arteries and arterioles in a state of partial constriction.
Without it, the vessels would fully dilate, and blood pressure would plummet. Adjusting this tone up or down is the primary way the VMC raises or lowers blood pressure.
2. Control of the Heart: A Dual System (Sympathetic & Parasympathetic)
The VMC influences the heart through two opposing pathways:
A. The Sympathetic "Accelerator" (Cardioacceleratory Center)
The lateral portions of the VMC function as the CARDIO-acceleratory center. When activated, it sends signals down the spinal cord to sympathetic nerves, which release norepinephrine onto the heart.
Increases Heart Rate (positive chronotropy)
Increases Contractility (positive inotropy), leading to a stronger pump and higher stroke volume.
Overall Goal: To increase Cardiac Output (CO).
B. The Parasympathetic "Brake" (Cardioinhibitory Center)
The medial portions of the VMC (often overlapping with the nucleus ambiguus) function as the CARDIOinhibitory center. It sends signals directly through the vagus nerves (Parasympathetic Nervous System - PSNS), which release acetylcholine onto the heart's pacemaker.
Decreases Heart Rate (negative chronotropy). Parasympathetic nerves have a minimal direct effect on ventricular contractility.
Overall Goal: To decrease Cardiac Output (CO).
How It All Integrates: The Baroreceptor Reflex Example
The true power of the VMC is how it coordinates these three functions simultaneously:
Scenario: A sudden increase in Blood Pressure (e.g., from a stress response)
Baroreceptors in the carotid sinuses and aorta detect high pressure and
baroreceptors increase their firing to the Sensory Area (NTS) of the medulla.
VMC Processing: The NTS signals the VMC to correct the high pressure.
Coordinated Output:
To Vessels: The Vasoconstrictor Area is inhibited. This reduces sympathetic vasomotor tone, causing vasodilation and a decrease in SVR.
To Heart: The Cardioacceleratory Center (lateral) is inhibited, reducing sympathetic drive to the heart.
To Heart: The Cardioinhibitory Center (medial) is stimulated, increasing parasympathetic (vagal) tone.
Result: The combined effect of a slower heart rate, reduced contractility, and widespread vasodilation brings blood pressure back down to normal.

baroreceptor firing
increase in blood pressure= increase firing
decrease in blood pressure= decrease in firing.
positive and negative chronotropy
chronotropy: heart rate
positive chronotopy: fast heart rate
negative chronotropy: negative heart rate
inotropy
inotropy: contractility
positive inotropy: makes the heart contract more forcefully (sympathetic nervous system and other factors)
negative inotropy: makes the heart contract less forcefully (parasympathetic nervous system, beta blockers and calcium channel blockers)
VMC Affects Vessel Function Via Neurotransmitters
which organ secretes EPI and NE?
what is the difference between b1 and b2 receptor?
Adrenal medulla secretes EPI and NE which constricts blood vessels via alpha adrenergic receptors
Epi/NE action on β1 receptors increases HR and SV
Epi dilates vessels through a potent β2 receptor response

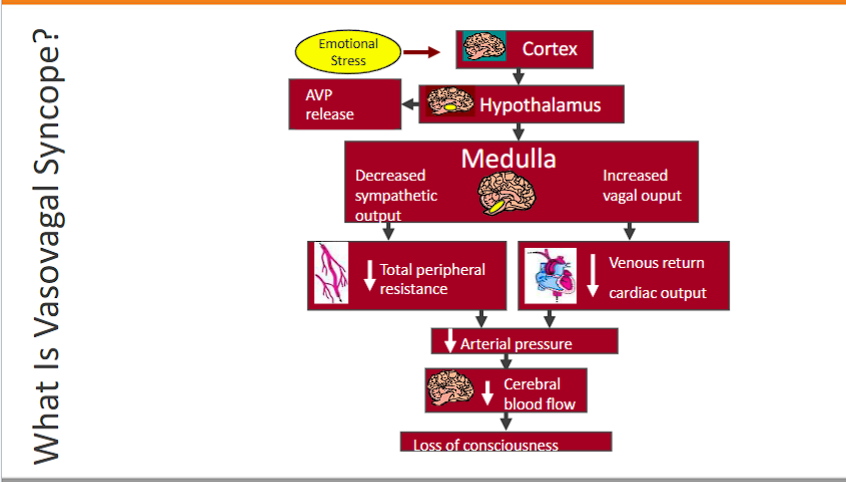
what is vasal syncope?
vasal syncope= faint
syncrope: “cutting off”
Vasovagal syncope is a sudden, reflex-mediated drop in heart rate and blood pressure, the drop in heart rate and blood pressure leads to a temporary reduction in cerebral blood flow and loss of consciousness. It is a neurologically triggered cardiovascular collapse.
1. The Trigger
Emotional Stress: Seeing blood, extreme fear, emotional distress.
Physical Stress: Standing for a long time, severe pain, dehydration, heat exposure.
2. Cortex The cortex interprets a stimulus as a threat and activates the emotional/stress centers that ultimately trigger the maladaptive reflex in the brainstem.
Hypothalamus (Sounds the Alarm & Activates Stress Pathways) AND releases AVP (vasopressin) to increase blood pressure, the release speed of AVP is slower than the drop in blood pressure, which is why the faint occurs.
Medulla: decrease sympathetic output and increases vagal tone, which leads to a decrease in TPR and decrease in venous return cardiac output, leading to a decrease in AP
decrease in arterial pressure leads to decrease in cerebral blood flow
decrease in cerebral blood flow leads to faint.
what is vasopressin?
water conservation (makes kidneys reabsorb water, triggered by high blood osmolarity)
vasoconstriction (raises blood pressure due to low blood volume)
Function | Mechanism | Primary Trigger |
|---|---|---|
Water Conservation (Anti-Diuresis) | Makes kidneys reabsorb water. | High Blood Osmolality ("Salty Blood") |
Vasoconstriction (Raises BP) | Squeezes blood vessels. | Low Blood Volume/Pressure (e.g., Bleeding) |
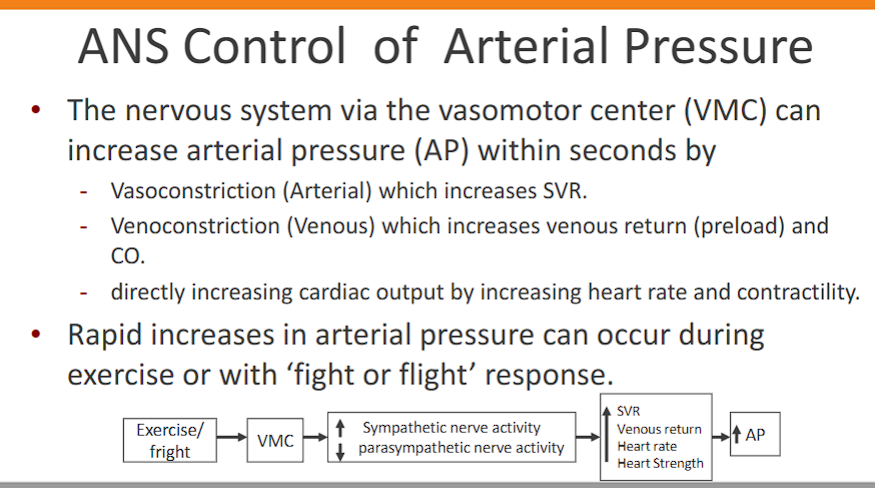
ANS control of arterial pressure (AP)?
the ANS uses the vasomotor center (VMC) is able to increase arterial pressure (AP) in seconds by:
Vasoconstriction (Arterial) which increases SVR
Venoconstriction (Venous) which increases venous return (preload) and CO
directly increasing cardiac output by increasing heart rate and contractility
Rapid increases in arterial pressure can occur during exercise or with ‘fight or flight’ response
what is the purpose of what you just learned about the ANS?
for the ANS to be able to control the arterial pressure (AP)
(the arterial baroreceptor reflex)
what is the relationship between baroreceptors and arterial pressure?
where are baroreceptors located?
how do baroreceptors control arterial pressure?
Important in short-term regulation of arterial pressure
baro: pressure
Reflex is initiated by stretch receptors called baroreceptors or pressoreceptors located in the walls of the large systemic arteries.
A rise in pressure stretches baroreceptors and causes them to send signals to the VMC
VMC sends feedback signals are sent via the autonomic nervous system (PSNS or SNS) to the circulation to reduce AP back to normal.

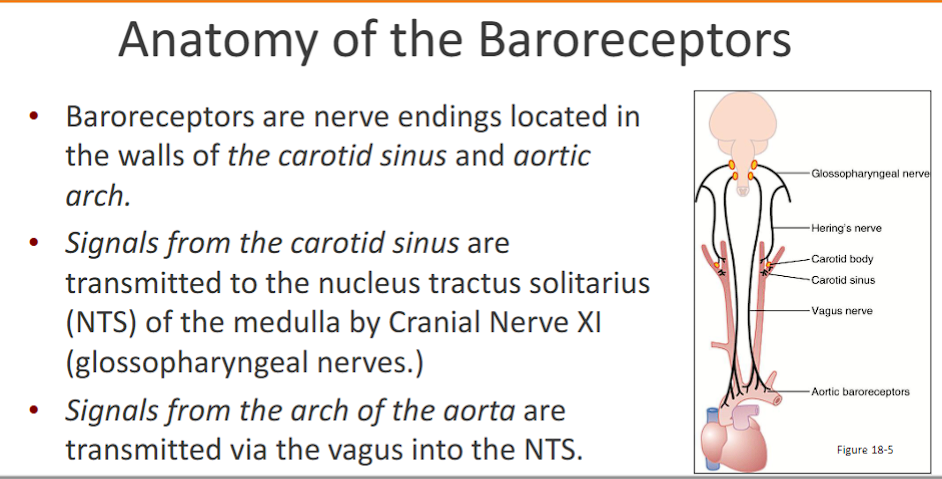
(anatomy of baroreceptors)
where are baroreceptors located?
where do the baroreceptors in the carotid sinus send signals? (where specifically in the medulla, because the medulla has the VMC)
where do the baroreceptors in the aortic arch send signals? (where specifically in the medulla, because the medulla has the VMC)
Baroreceptors are nerve endings located in the walls of the carotid sinus and aortic arch
Signals from the carotid sinus are transmitted to the nucleus tractus solitarius (NTS) of the medulla by Cranial Nerve XI (glossopharyngeal nerves.) “tonge throat nerve”
Signals from the arch of the aorta are transmitted via the vagus into the NTS (Nucleus of the Tractus Solitarius)
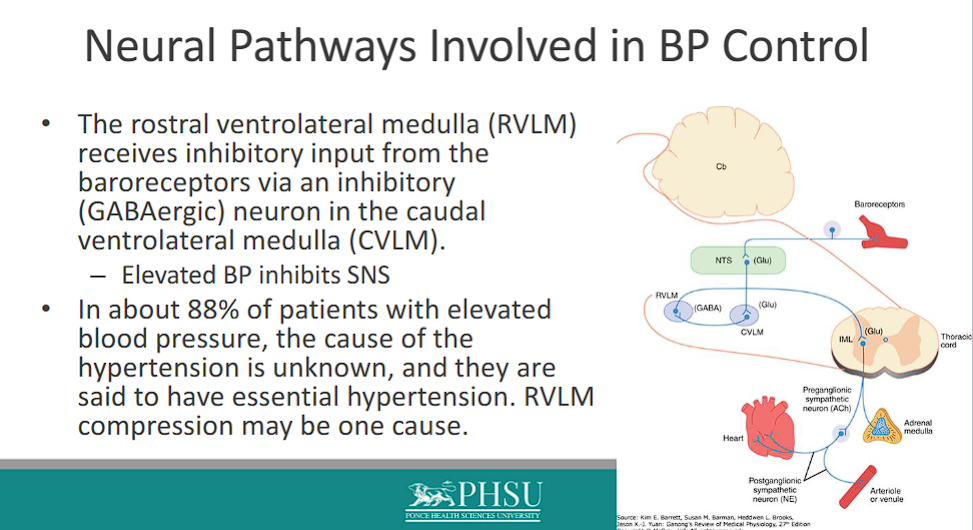
(Neural Pathways Involved in BP Control)
The rostral ventrolateral medulla (RVLM) receives inhibitory input from the baroreceptors via an inhibitory (GABAergic) neuron in the caudal ventrolateral medulla (CVLM)
High BP → Baroreceptors ↑ firing → NTS → Excites CVLM → CVLM Inhibits RVLM (via GABA) → ↓ Sympathetic Outflow → Vasodilation & ↓ Heart Rate → BP RETURNS TO NORMAL
RVLM (Rostral Ventrolateral Medulla): The "ON Switch" for blood pressure. It sends excitatory signals to the sympathetic nerves that constrict blood vessels and increase heart rate. If the RVLM is active, blood pressure goes up.
CVLM (Caudal Ventrolateral Medulla): The "OFF Switch" for the RVLM. Its job is to inhibit the RVLM.
NTS (Nucleus of the Tractus Solitarius): The "Sensory Hub." It receives all the sensory input about blood pressure from the baroreceptors.
– Elevated BP inhibits SNS
In about 88% of patients with elevated blood pressure, the cause of the hypertension is unknown, and they are said to have essential hypertension. RVLM compression may be one cause
what does an elevated blood pressure do the SNS?
– Elevated BP inhibits SNS

(How Do the Baroreceptors Respond to Changes in Arterial Pressure?)
what do baroreceptors do?
at what arterial pressure are baroreceptors most sensitive at?
Carotid sinus baroreceptors respond to pressures between 60 – 180 mm Hg.
• Baroreceptors respond to changes in arterial pressure.
• Baroreceptor reflex is most sensitive at a pressure of 100 mm Hg.
• As pressure increases the number of impulses from carotid sinus increases which results in
(1) inhibition of the vasoconstrictor (SNS)
(2) activation of the vagal center (PSNS)
Constrict Carotids (constriction= less pressure= less stretcing)→ ↓ Pressure in Carotid Sinus → ↓ Baroreceptor Firing → Medulla (NTS/VMC) → ↑ Sympathetic Outflow & ↓ Parasympathetic Outflow → ↑ Heart Rate & ↑ Vasoconstriction → ↑ Arterial Pressure
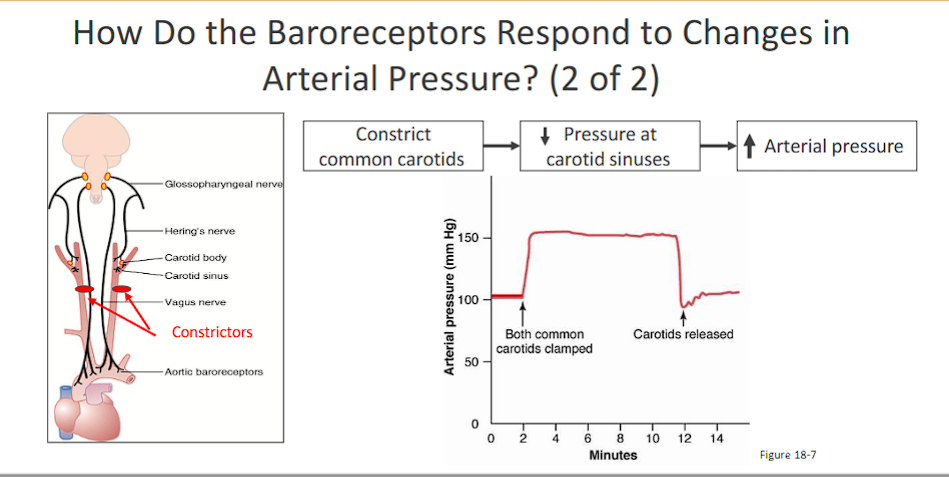
what does vagal center mean?
PSNS
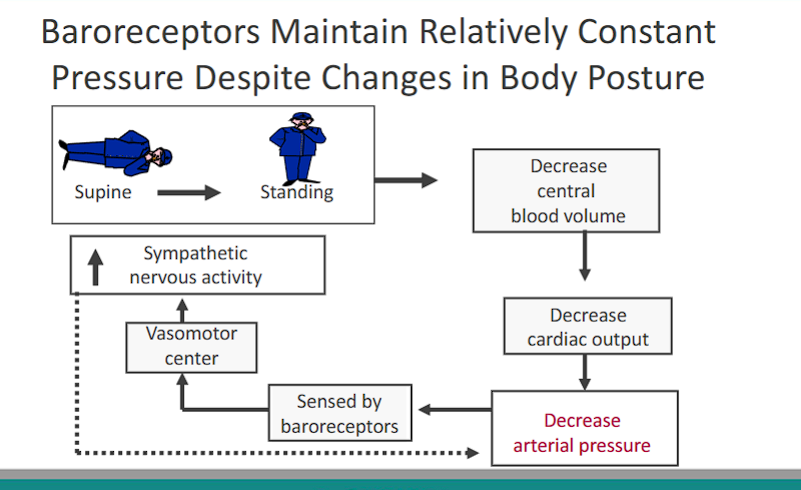
what is the relationship between baroreceptors and body posture
Baroreceptors Maintain Relatively Constant Pressure Despite Changes in Body Posture
Carotid and Aortic Chemoreceptors
what are the three things chemoreceptors detect?
where are chemoreceptors located?
what does activation of chemoreceptors cause?
when are chemoreceptors stimulated?
Chemo: chemistry
Chemoreceptors are chemosensitive cells sensitive to low oxygen, CO2 excess, or low pH.
Chemoreceptors are located in carotid bodies near the carotid bifurcation and on the arch of the aorta
Activation of chemosensitive receptors results in excitation of the vasomotor center = increased sympathetic nervous system.
Chemoreceptors are not stimulated until pressure falls below 80 mm Hg

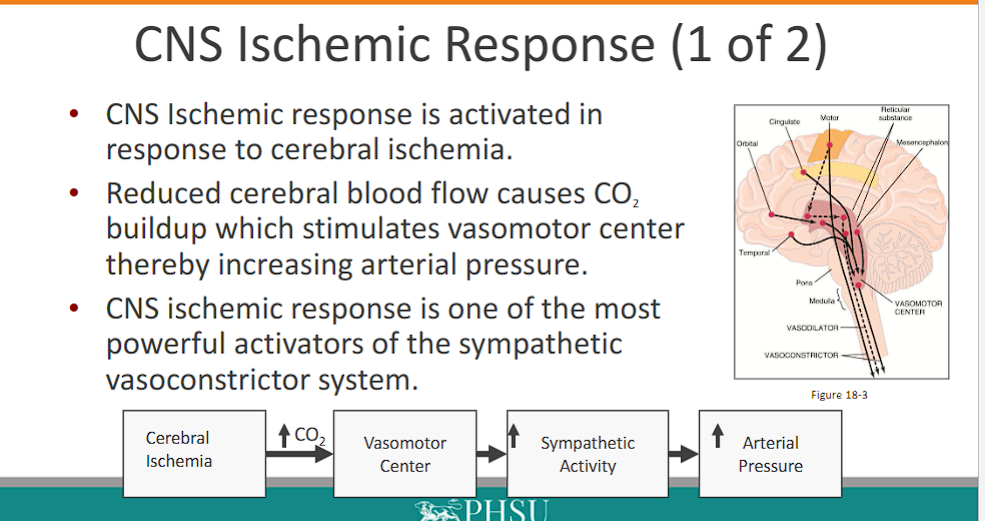
CNS Ischemic Response
Cerebral: From Latin cerebrum, meaning "brain."
Ischemia: From Greek isch- (ἴσχω, ischō), meaning "to restrict" or "to hold back," and -emia (αἷμα, haima), meaning "blood."
translation: "A restriction of blood to the brain."
CNS Ischemic response is activated in response to cerebral ischemia
the brain is a metabolic powerhouse that relies on blood for the export of metabolic byproduct of CO2. however, when blood flow is reduced (cerebral ischemia) but CO2 is still being produced, CO2 buildup occurs.
Reduced cerebral blood flow causes CO2 buildup which stimulates chemoreceptors, which stimulates vasomotor center thereby increasing arterial pressure (and hyperventilation)
CNS ischemic response is one of the most powerful activators of the sympathetic vasoconstrictor system.
CNS Ischemic Response (2 of 2)
when is the CNS ischemic response activated?
when is the greatest CNS ischemic response?
CNS ischemic response is not activated until pressure falls below 60 mm Hg; greatest activation occurs at pressures of 15–20 mm Hg.
Cushing reaction is a special type of CNS ischemic response
Prolonged CNS ischemia has a depressant effect on the vasomotor center.

Cushing Reflex
"Cushing Reflex" does not describe the mechanism in its name. Instead, it honors the individual who first characterized the phenomenon.
Cushing reaction is a special type of CNS ischemic response.
The Cushing Reflex is a specific triad of clinical signs that occur when Intracranial Pressure (ICP) rises to a critical level, compressing the brain and its blood vessels. The classic triad is:
Hypertension (High Systemic Blood Pressure)
Bradycardia (Slow Heart Rate)
Irregular Respiration
When ICP increases, it compresses the brainstem, which triggers SNS to release norepinephrine leading
to:
– Vasoconstriction, increasing blood pressure
– decreased heart rate (due to baroreceptors detecting increase in blood pressure)
– Release of renin, leading to further elevation of BP
Causes include brain tumors, intracranial hemorrhage, stroke, hydrocephalus, and space-occupying lesion

Factors affecting heart rate

(Atrial and Pulmonary Artery Reflexes)
-Low pressure receptors in atria and pulmonary arteries minimize arterial pressure changes in response to changes in blood volume.
-Increases in blood volume activate low pressure receptors which in turn lower arterial pressure
low pressure receptors want to decrease AP.
-Activation of low-pressure receptors gets rid of Na and water by:
• decreasing rate of antidiuretic hormone
• Increasing glomerular filtration rate.
• decreasing Na reabsorption
blood volume increases
atrial stretch increases
low pressure receptors activate
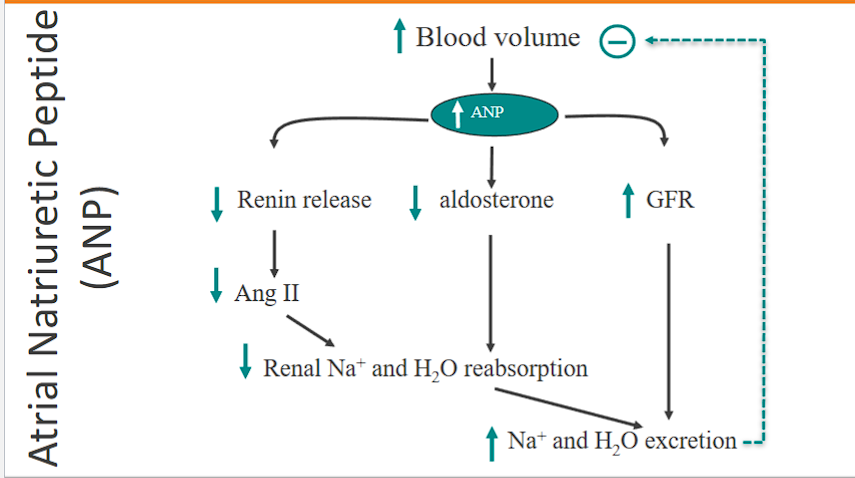
Renal sympathetic activity DECREASES and ANP INCREASES (anp is the OPPOSITE of vasopressin, ADP is the same as vasopressin)
decrease in blood volume achieved.

ANP, ADP, vasopressin
anp is the opposite of adh (vasopressin)
adh is antidieuretic, so it makes you retain water and sodium
anp (Atrial Natriuretic Peptide) is the opposite of adh, so it makes you lose water and sodium.
natriuretic= sodium passing through urine.
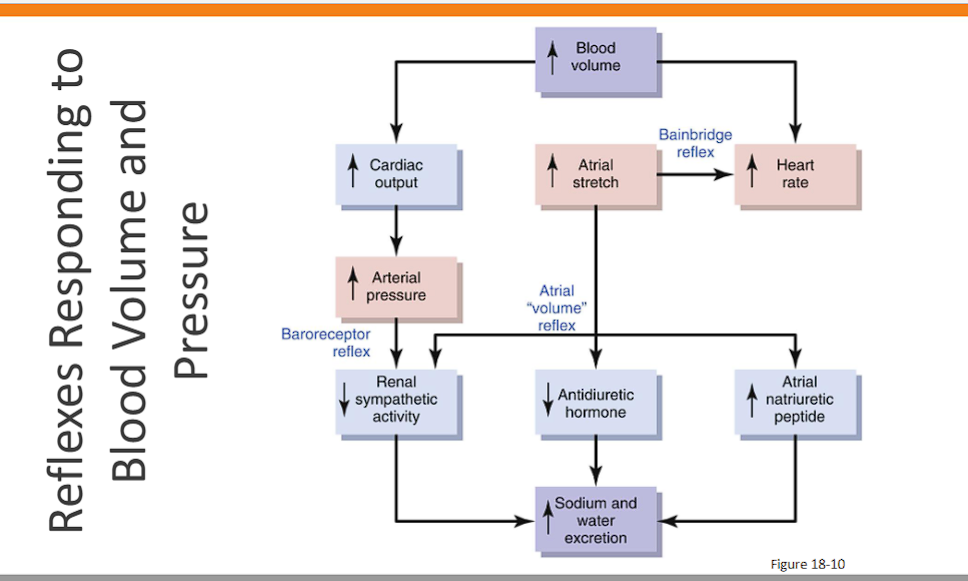
Bainbridge Reflex
bainbridge: the scientist
Prevents damming of blood in veins atria and pulmonary circulation, preventing congestion of blood.
The Bainbridge Reflex is a cardiovascular response that helps the heart manage a sudden increase in blood volume returning to the heart.
pressure increase (caused by sudden increase in blood volume due to blood transfusion, IV or exercise)
atrial stretch occurs
LOW PRESSURE BARORECEPTORS activated
stretch signals travel to vagal afferent nerves
VMC interprets signals
Efferent Response: The VMC responds by:
Withdrawing Parasympathetic (Vagal) Tone: This is the primary mechanism, leading to an increase in heart rate.
Increasing Sympathetic Outflow to the Heart: This can also contribute to increased heart rate and may enhance contractility
increase in contractility and heart rate to make the heart beat faster to move the extra blood.
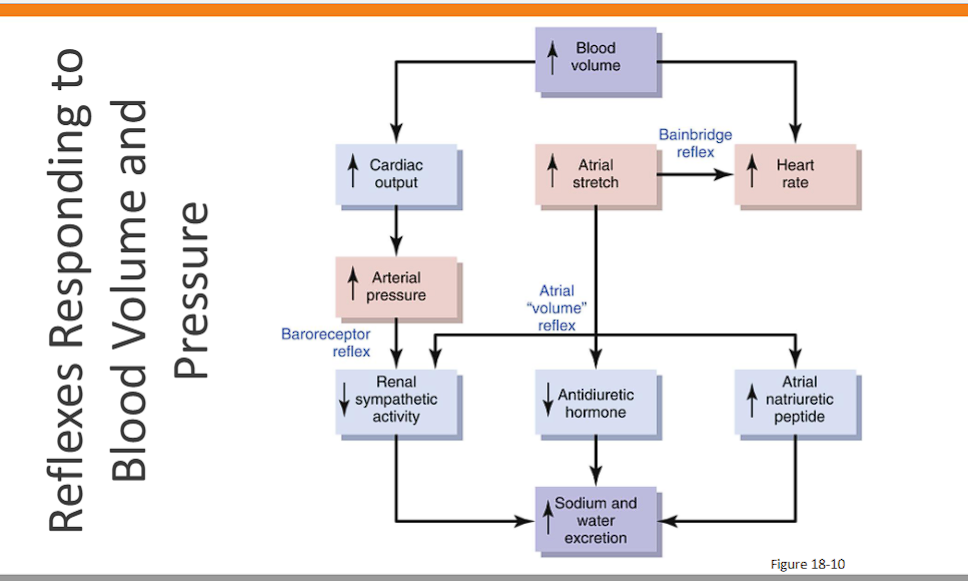
(Reflexes Responding to Blood Volume and Pressure)
baroreceptor reflex
baroreceptor reflex: increase in blood volume increases AP, more baroreceptors fire to lower the blood pressure, decreasing sympathetic renal activity to increase water and sodium excretion.
atrial volume reflex: The term "Atrial Volume Reflex" is essentially another name for the Bainbridge Reflex that you just described.
Renal Regulation of ECFV/BP
what is the relationship between ECFV and BP? (makes sense)
Plays a dominant role in long-term pressure control
direct relationship: As extracellular fluid volume increases arterial pressure increases.
The increase in arterial pressure causes the kidneys to lose Na and water which returns extracellular fluid volume (ECFV) to normal.

Pressure Natriuresis and Diuresis
The effect of pressure to increase water excretion is called pressure diuresis.
The effect of pressure to increase Na excretion is called pressure natriuresis.
if AP increases, these increase.
if AP decreases, these decrease
Together, they form the Pressure-Natriuresis-Diuresis Mechanism.

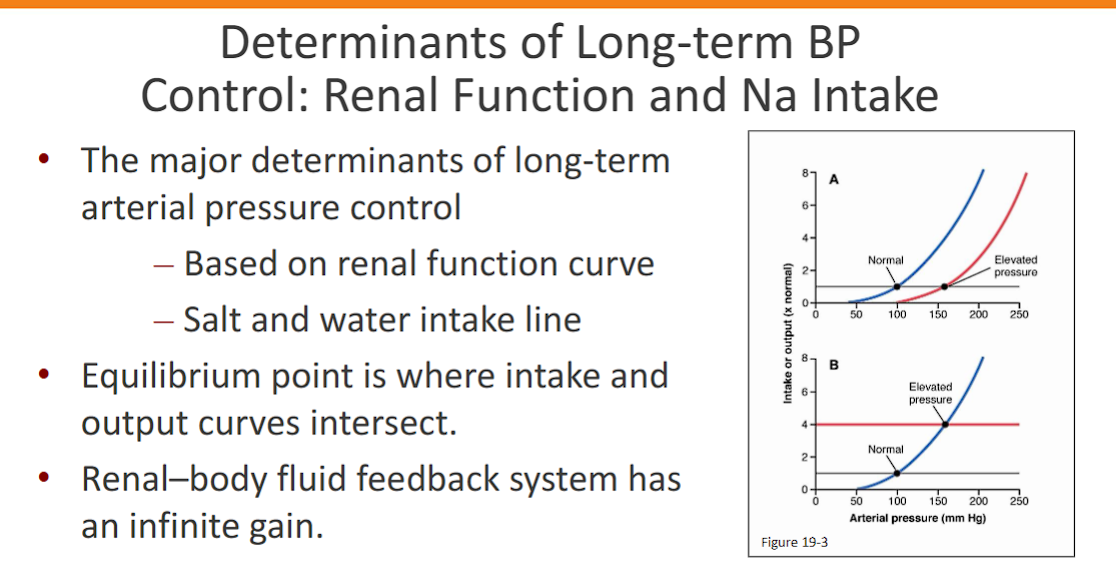
Determinants of Long-term BP Control: Renal Function and Na Intake
The major determinants of long-term
arterial pressure control
– Based on renal function curve
– Salt and water intake line
Equilibrium point is where intake and
output curves intersect.
• Renal–body fluid feedback system has
an infinite gain
What it is: This curve represents the kidney's response to pressure—the Pressure-Natriuresis-Diuresis mechanism we just discussed.
Shape: It is very steep. A small increase in arterial pressure causes a large increase in sodium and water excretion by the kidneys.
Meaning: The kidneys are exquisitely sensitive to pressure. If pressure rises even slightly, the kidneys will rapidly dump salt and water until the pressure is forced back down.
short term and long term blood pressure control.
baroreceptors and chemoreceptors are short term, renal function and na intake are long term blood pressure control
Failure of SVR to Elevate Long-term B
-Changes in SVR (vasoconstriction vasodilation) do not affect long- term arterial pressure level
-therefore, one must alter the renal function curve in order to have long-term changes in arterial pressure.
-Changing renal vascular resistance does not lead to long-term changes in BP.
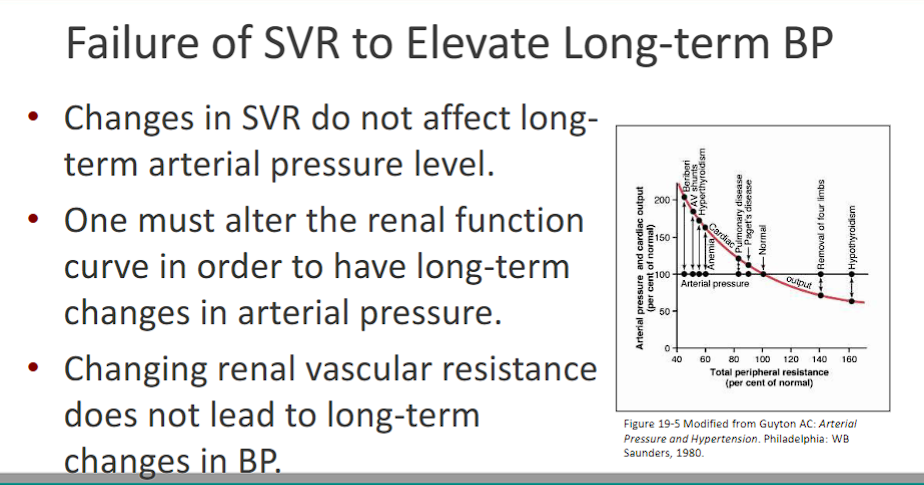
MAKE SURE THE STATEMENT IS CORRECT
CHANGES IN RENAL VASCULAR RESISTANCE DOES NOT LEAD TO LONG TERM CHANGES IN BP.
RENAL NA AND WATER EXCRETION LEAD TO LONG TERM CHANGES IN BLOOD PRESSURE.
Sodium Is a Major Determinant of ECFV
-As Na intake is increased, Na stimulates drinking, increased Na concentration stimulates thirst and ADH secretion.
-Changes in Na intake lead to changes in extracellular fluid volume (ECFV).
-ECFV is determined by the balance of Na intake and output.

what does high blood osmolarity mean?
high salt
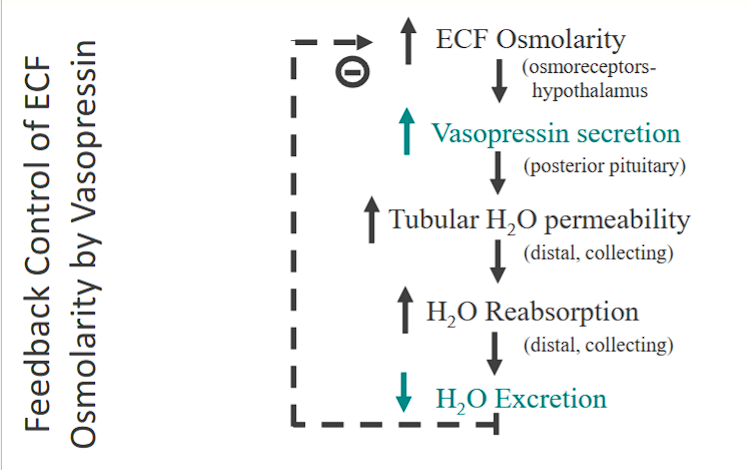
Feedback Control of ECF Osmolarity by Vasopressin
The Pathway (as you outlined):
Stimulus: ↑ ECF Osmolarity (high salt) (e.g., from dehydration or high salt intake).
Sensor: Osmoreceptors in the hypothalamus.
Control Center: Hypothalamus and Posterior Pituitary.
Effector/Hormone: ↑ Vasopressin (ADH) Secretion.
Action on Kidney: ↑ Tubular H₂O Permeability in the distal tubule and collecting duct.
Effect: ↑ H₂O Reabsorption, ↓ H₂O Excretion.
Final Result: ECF Osmolarity returns to normal.
ANP
Renin-Angiotensin System
decreased arterial pressure
Renin release from the juxtaglomerular (JG) cells in the kidney is stimulated by three primary factors (low blood pressure in afferent, low sodium delivery, increase in sympathetic nervous system) in response to low blood pressure
Renin (from kidney) acts on Angiotensinogen (a protein made by the liver) to form Angiotensin I (a relatively inactive decapeptide).
Angiotensin-Converting Enzyme (ACE), located predominantly on the surface of endothelial cells in the lungs, converts Angiotensin I into the highly active Angiotensin II (an octapeptide)
3. The Powerful Effects of Angiotensin II (AII)
Angiotensin II is the primary effector of the system and works through several mechanisms to raise blood pressure:Potent Vasoconstriction: Causes direct, powerful constriction of arterioles throughout the body, dramatically increasing Systemic Vascular Resistance (SVR).
Stimulates Aldosterone Release: Acts on the adrenal cortex to release aldosterone. Aldosterone tells the kidneys to reabsorb more sodium (and consequently, water), which increases blood volume.
Stimulates ADH (Vasopressin) Release: Promotes water reabsorption by the kidneys to further increase blood volume.
Stimulates Thirst: In the brain, to promote fluid intake.
Increases Sympathetic Outflow: Amplifies the activity of the sympathetic nervous system

what are the three things angiotensin II does?
Potent Vasoconstriction to increase SVR
Stimulates Aldosterone Release to increase sodium retention to increase blood volume.
Stimulates ADH (Vasopressin) Release: increases water retention to increase blood volume.
Stimulates Thirst: In the brain, to promote fluid intake
Increases Sympathetic Outflow: Amplifies the activity of the sympathetic nervous system.

what is the GOAL of RAS?
The RAS is activated to correct low blood volume and low blood pressure. It does this by:
Constricting blood vessels (↑ SVR)
Conserving salt and water (↑ Blood Volume)
According to the equation Arterial Pressure = Cardiac Output × Systemic Vascular Resistance, the RAS powerfully increases both components on the right side of the equation to raise arterial pressure.
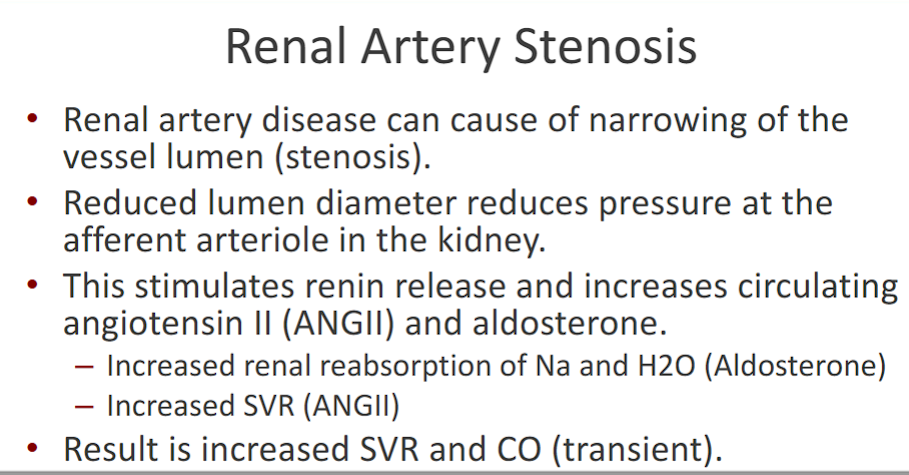
Renal Artery Stenosis
-narrowing of the AFFERENT renal artery
-Reduced lumen diameter reduces pressure at the afferent arteriole in the kidney
the reduced pressure due to the narrowing: stimulates renin release and increases circulating angiotensin II (ANGII) and aldosterone.
– Increased renal reabsorption of Na and H2O (Aldosterone)
– Increased SVR (ANGII)
• Result is increased SVR and CO (transient)
Heart Failure
1. The Initial Insult ("Significant Event")
Myocardial infarction (most common), hypertension, valvular heart disease, cardiomyopathy, etc.
Effect: Damaged or overworked heart muscle leads to impaired pumping capacity and a severely reduced Cardiac Output (CO).
2. Compensatory Neurohormonal Activation
The body misinterprets the low CO as a sign of low blood volume/pressure and activates its classic "rescue" systems:
Sympathetic Nervous System (SNS): The first and fastest responder.
Goal: Increase CO and BP.
Effects: Tachycardia, increased contractility, and systemic vasoconstriction.
Renin-Angiotensin-Aldosterone System (RAAS): A slower but powerful hormonal response.
Goal: Increase blood volume and BP.
Effects: Angiotensin II causes potent vasoconstriction and thirst. Aldosterone causes sodium and water retention.
3. The Double-Edged Sword: Short-Term Gain vs. Long-Term Pain
Short-Term Restoration of Homeostasis: Initially, these responses are beneficial. They help maintain blood pressure and perfusion to vital organs by:
Supporting a failing heart rate and contractility.
Maintaining central blood volume.
Sustained Deleterious Effects: When these systems remain chronically active, they become toxic to the cardiovascular system:
Tachycardia & High Contractility: Increase myocardial oxygen demand, straining the failing heart and potentially leading to arrhythmias.
Vasoconstriction: Increases the afterload (the pressure the heart must pump against). This further reduces stroke volume and CO, worsening the heart's efficiency.
Sodium & Water Retention: Increases the preload (the volume of blood the heart must pump). In a stiff, failing heart, this leads to elevated filling pressures, causing pulmonary congestion (shortness of breath) and peripheral edema (swelling).
The Result: A vicious cycle where the very mechanisms meant to save the system end up accelerating its decline.

Heart Failure Treatment (going against the body’s mechanisms for treatment of the heart)
The Basis of Modern Therapy: Breaking the Cycle
As you correctly stated, antagonizing these neurohormonal systems is the cornerstone of heart failure management.
Beta-Blockers: Antagonize the SNS. They slow heart rate, reduce contractility (reducing oxygen demand), and have anti-arrhythmic effects.
ACE Inhibitors (or ARBs): Antagonize the RAAS. They prevent the formation/action of Angiotensin II, leading to vasodilation (↓ afterload), reduced aldosterone (↓ fluid retention), and prevention of harmful cardiac remodeling.
Mineralocorticoid Receptor Antagonists (e.g., Spironolactone): Directly block aldosterone, further promoting diuresis and preventing fibrosis.
Diuretics: While not direct neurohormonal blockers, they are used to counteract the fluid-retaining effects of the activated RAAS and SNS, relieving symptoms of congestion.
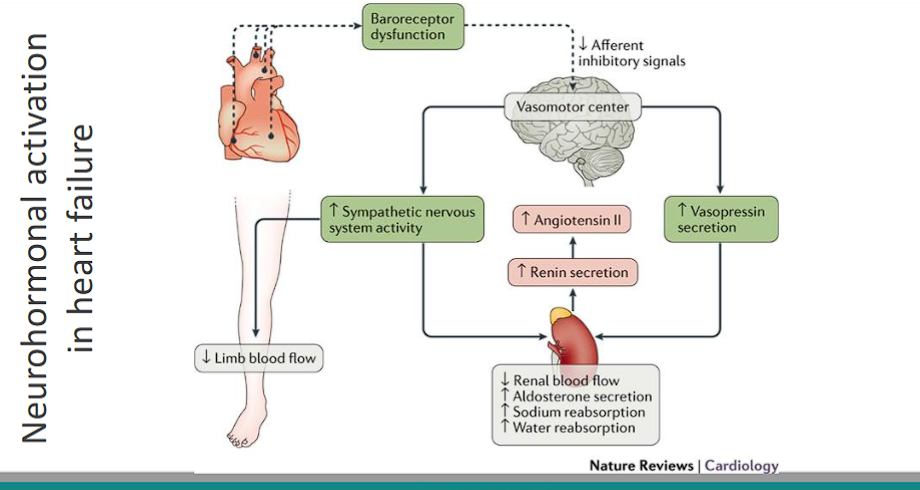
Neurohormonal activation in heart failure
-sympathetic nervous system norepinephrine (neurotransmitter)
-hormone (RAAS, vasopressin)
heart dysfunction
baroreceptor dysfunction
afferent inhibitory signals sent to VMC
VMC increases sympathetic output and increases vasopressin secretion
Kidney increases renin → angiotensin II
increased angiotensin II leads to DECREASED RENAL BLOOD FLOW, increased ALDOSTERONE, INCREASED SODIUM reabsorption and INCREASED water reabsorption.
(Hypertension Is Classified as)
Prehypertension
Hypertension
Stage 1 hypertension
Stage 2 hypertension
Normal blood pressure: < 120/80 mm Hg
• Prehypertension: 120–139/80–89 mm Hg
• Hypertension: greater than 140/90 mm Hg
• Stage 1 hypertension:140–159/90–99 mm Hg
• Stage 2 hypertension: 160 or greater/100 or greater mm Hg

Primary or Essential Hypertension (weight gain)
90% of hypertensive patients
• Mild form of hypertension, slow progression
• Cause is unknown but most likely related to weight gain.
• 2/3 of essential hypertensives are overweight

Treating Hypertension (you already went over this)
Drugs used to treat hypertension include:
• Angiotensin converting enzyme (ACE) inhibitors
• Angiotensin receptor blockers (ARBs)
• Diuretics
• Beta-blockers
• Calcium channel blockers

Describe the origin and distribution of sympathetic and parasympathetic nerves to the heart and circulation. (learning objective)
Here is a detailed description of the origin and distribution of sympathetic and parasympathetic nerves to the heart and circulation.
Overview
The autonomic nervous system regulates cardiac and vascular function through two opposing branches: the Sympathetic and Parasympathetic systems. Their general effects are:
Sympathetic ("Fight-or-Flight"): Increases heart rate, contractility, and causes vasoconstriction in most vascular beds to elevate blood pressure and redirect blood flow.
Parasympathetic ("Rest-and-Digest"): Decreases heart rate and contractility (with a much stronger effect on rate). It has minimal direct innervation of blood vessels, with key exceptions.
Sympathetic Innervation
1. Origin: The Thoracolumbar Outflow
Sympathetic nerves to the heart and circulation originate from the intermediolateral (IML) cell column (the lateral horn) of the spinal cord.
For the Heart: Preganglionic neurons arise from spinal cord segments T1 to T4/T5.
For the Vasculature: Preganglionic neurons for blood vessels throughout the body arise from segments T1 to L2/L3. This is why it's called the thoracolumbar outflow.
2. Pathway and Distribution
The pathway involves a two-neuron chain: a preganglionic neuron and a postganglionic neuron.
A. To the Heart:
Preganglionic Fibers: The axons from T1-T4 neurons leave the spinal cord via the ventral roots and enter the sympathetic chain (a bundle of ganglia running parallel to the spine) via white rami communicantes.
Synapse: These fibers ascend within the chain and synapse with postganglionic neuronal cell bodies located in the cervical and upper thoracic ganglia (specifically the superior, middle, and stellate ganglia).
Postganglionic Fibers: From these ganglia, the postganglionic fibers form several cardiac nerves (superior, middle, and inferior cervical cardiac nerves, and thoracic cardiac nerves).
Innervation: These cardiac nerves proceed to the cardiac plexus (a network of nerves at the base of the heart) and then distribute widely to:
Sinoatrial (SA) Node: To increase heart rate.
Atrioventricular (AV) Node: To increase conduction velocity.
Atrial and Ventricular Myocardium: To increase the force of contraction.
B. To the Blood Vessels (Vascularure):
Preganglionic Fibers: The axons from T1-L2 neurons enter the sympathetic chain.
Synapse: They can synapse at the same level, ascend/descend to synapse at a different level, or pass through the chain without synapsing to form splanchnic nerves that synapse in prevertebral ganglia (e.g., celiac, superior mesenteric).
Postganglionic Fibers:
For limbs and body wall: From the chain ganglia, postganglionic fibers travel via gray rami communicantes to join spinal nerves, which then distribute to blood vessels (especially arterioles) in the skin, skeletal muscle, and other somatic tissues.
For viscera: From the prevertebral ganglia, postganglionic fibers follow arteries to reach the blood vessels of the abdominal and pelvic organs.
Effect: The primary effect is vasoconstriction via the release of norepinephrine. The only major exception is in skeletal muscle, where sympathetic cholinergic fibers can cause vasodilation during stress (the "get-ready" response).
Parasympathetic Innervation
1. Origin: The Craniosacral Outflow
Parasympathetic nerves originate from the brainstem and the sacral spinal cord. For the heart and thoracic vasculature, the source is exclusively cranial, specifically the Vagus Nerves (Cranial Nerve X).
Preganglionic neuronal cell bodies are located in the dorsal motor nucleus of the vagus and the nucleus ambiguus in the medulla oblongata.
2. Pathway and Distribution
The pathway also uses a two-neuron chain, but the ganglia are located very close to or within the target organ.
A. To the Heart:
Preganglionic Fibers: The axons travel down within the vagus nerves (left and right) through the neck and thorax.
Synapse: These long preganglionic fibers do not synapse until they reach the cardiac plexus. Here, they synapse with postganglionic neuronal cell bodies located in small ganglia embedded in the plexus or within the walls of the atria themselves (e.g., the ganglia subdividing into the SA and AV nodes).
Postganglionic Fibers: These are very short and project directly to their target structures:
SA Node: To decrease heart rate (strong effect).
AV Node: To decrease conduction velocity (strong effect).
Atrial Myocardium: To decrease the force of contraction (moderate effect).
Ventricular Myocardium: Very sparse innervation; the parasympathetic system has a minimal direct effect on ventricular contractility.
B. To the Blood Vessels (Vascularure):
The parasympathetic system has very limited and specific distribution to blood vessels. It does not provide widespread innervation like the sympathetic system.
Regions Innervated: It primarily innervates blood vessels of certain exocrine glands (salivary, lacrimal, gastric) to promote vasodilation and secretion, and vessels of the external genitalia for erection (via pelvic splanchnic nerves, S2-S4).
Pathway: Preganglionic fibers in the vagus nerve synapse in ganglia near the specific organs (e.g., otic, pterygopalatine), and very short postganglionic fibers release Acetylcholine (ACh), which triggers vasodilation indirectly, often through the release of Nitric Oxide (NO).
General Systemic Circulation: Most arterioles and veins do not receive parasympathetic innervation. Systemic blood pressure is primarily regulated by the sympathetic nervous system.
Summary Table
Feature | Sympathetic Nerves | Parasympathetic Nerves |
|---|---|---|
Origin (Outflow) | Thoracolumbar (T1-L2) | Cranial (Vagus Nerve, CN X) & Sacral |
Preganglionic Neurotransmitter | Acetylcholine (ACh) | Acetylcholine (ACh) |
Postganglionic Neurotransmitter | Norepinephrine (NE) | Acetylcholine (ACh) |
Ganglion Location | Near Spine (Paravertebral/Prevertebral) | Near or within the Heart |
Innervation of Heart | SA Node, AV Node, Atria, Ventricles | SA Node, AV Node, Atria (ventricles sparse) |
Cardiac Effect | ↑ HR (Chronotropy), ↑ Contractility (Inotropy) | ↓ HR (Chronotropy), ↓ Atrial Contractility |
Innervation of Vasculature | Widespread: Skin, Gut, Skeletal Muscle, etc. | Limited & Specific: Glands, Genitalia |
Vascular Effect | Vasoconstriction (primarily); Vasodilation in skeletal muscle | Vasodilation (in specific beds) |
autonomic nervous system (overview)
effect of sympathetic nervous system on heart rate, contractility, vasoconstriction and vasodilation
effect of parasympathetic nervous system on heart rate, contractility, vasoconstriction and vasodilation
The autonomic nervous system regulates cardiac and vascular function through two opposing branches: the Sympathetic and Parasympathetic systems. Their general effects are:
Sympathetic ("Fight-or-Flight"): Increases heart rate, increases contractility, and causes vasoconstriction in most vascular beds to elevate blood pressure and redirect blood flow.
Parasympathetic ("Rest-and-Digest"): Decreases heart rate and decreases contractility (with a much stronger effect on rate). It has minimal direct innervation of blood vessels, with key exceptions.
Sympathetic Innervation
origin (intermediolateral cell column)
pathway
synapse
innervation
parasympathetic innervation
Sympathetic Innervation
1. Origin: The Thoracolumbar Outflow
Sympathetic nerves to the heart and circulation originate from the intermediolateral (IML) cell column (the lateral dshorn) of the spinal cord.
For the Heart: Preganglionic neurons arise from spinal cord segments T1 to T4/T5.
For the Vasculature: Preganglionic neurons for blood vessels throughout the body arise from segments T1 to L2/L3. This is why it's called the thoracolumbar outflow.
2. Pathway and Distribution
The pathway involves a two-neuron chain: a preganglionic neuron and a postganglionic neuron.
A. To the Heart:
Preganglionic Fibers: The axons from T1-T4 neurons leave the spinal cord via the ventral roots and enter the sympathetic chain (a bundle of ganglia running parallel to the spine) via white rami communicantes.
Synapse: These fibers ascend within the chain and synapse with postganglionic neuronal cell bodies located in the cervical and upper thoracic ganglia (specifically the superior, middle, and stellate ganglia).
Postganglionic Fibers: From these ganglia, the postganglionic fibers form several cardiac nerves (superior, middle, and inferior cervical cardiac nerves, and thoracic cardiac nerves).
Innervation: These cardiac nerves proceed to the cardiac plexus (a network of nerves at the base of the heart) and then distribute widely to:
Sinoatrial (SA) Node: To increase heart rate.
Atrioventricular (AV) Node: To increase conduction velocity.
Atrial and Ventricular Myocardium: To increase the force of contraction.
B. To the Blood Vessels (Vascularure):
Preganglionic Fibers: The axons from T1-L2 neurons enter the sympathetic chain.
Synapse: They can synapse at the same level, ascend/descend to synapse at a different level, or pass through the chain without synapsing to form splanchnic nerves that synapse in prevertebral ganglia (e.g., celiac, superior mesenteric).
Postganglionic Fibers:
For limbs and body wall: From the chain ganglia, postganglionic fibers travel via gray rami communicantes to join spinal nerves, which then distribute to blood vessels (especially arterioles) in the skin, skeletal muscle, and other somatic tissues.
For viscera: From the prevertebral ganglia, postganglionic fibers follow arteries to reach the blood vessels of the abdominal and pelvic organs.
Effect: The primary effect is vasoconstriction via the release of norepinephrine. The only major exception is in skeletal muscle, where sympathetic cholinergic fibers can cause vasodilation during stress (the "get-ready" response).
Parasympathetic innervation
origin
pathway and distribution to the heart
pathway and distribution to the blood vessels.
Parasympathetic Innervation
1. Origin: The Craniosacral Outflow
Parasympathetic nerves originate from the brainstem and the sacral spinal cord. For the heart and thoracic vasculature, the source is exclusively cranial, specifically the Vagus Nerves (Cranial Nerve X).
Preganglionic neuronal cell bodies are located in the dorsal motor nucleus of the vagus and the nucleus ambiguus in the medulla oblongata.
2. Pathway and Distribution
The pathway also uses a two-neuron chain, but the ganglia are located very close to or within the target organ.
A. To the Heart:
Preganglionic Fibers: The axons travel down within the vagus nerves (left and right) through the neck and thorax.
Synapse: These long preganglionic fibers do not synapse until they reach the cardiac plexus. Here, they synapse with postganglionic neuronal cell bodies located in small ganglia embedded in the plexus or within the walls of the atria themselves (e.g., the ganglia subdividing into the SA and AV nodes).
Postganglionic Fibers: These are very short and project directly to their target structures:
SA Node: To decrease heart rate (strong effect).
AV Node: To decrease conduction velocity (strong effect).
Atrial Myocardium: To decrease the force of contraction (moderate effect).
Ventricular Myocardium: Very sparse innervation; the parasympathetic system has a minimal direct effect on ventricular contractility.
B. To the Blood Vessels (Vascularure):
The parasympathetic system has very limited and specific distribution to blood vessels. It does not provide widespread innervation like the sympathetic system.
Regions Innervated: It primarily innervates blood vessels of certain exocrine glands (salivary, lacrimal, gastric) to promote vasodilation and secretion, and vessels of the external genitalia for erection (via pelvic splanchnic nerves, S2-S4).
Pathway: Preganglionic fibers in the vagus nerve synapse in ganglia near the specific organs (e.g., otic, pterygopalatine), and very short postganglionic fibers release Acetylcholine (ACh), which triggers vasodilation indirectly, often through the release of Nitric Oxide (NO).
General Systemic Circulation: Most arterioles and veins do not receive parasympathetic innervation. Systemic blood pressure is primarily regulated by the sympathetic nervous system.
Know the location and function of alpha- and beta-adrenoceptors and muscarinic receptors in the heart and blood vessels.
Knowing the location and function of these receptors is fundamental to understanding cardiovascular physiology and pharmacology.
Here is a detailed breakdown of the location and function of alpha-adrenoceptors, beta-adrenoceptors, and muscarinic receptors in the heart and blood vessels.
Overview of Neurotransmitters
Sympathetic Nerves release Norepinephrine (and epinephrine from the adrenal medulla), which acts on alpha and beta-adrenoceptors.
Parasympathetic Nerves release Acetylcholine (ACh), which acts on muscarinic receptors.
1. Alpha-Adrenoceptors (α-receptors)
Alpha-receptors are primarily involved in vasoconstriction (narrowing of blood vessels).
Location:
Blood Vessels: Predominantly located on vascular smooth muscle of most arterioles and veins.
α₁-receptors: Mainly post-synaptic, on the target organ (the blood vessel).
α₂-receptors: Primarily pre-synaptic (on the nerve terminal) where they act as auto-receptors to inhibit further norepinephrine release. Some are also post-synaptic on vascular smooth muscle.
Heart: Present in much lower density. Their role is less direct.
Function:
In Blood Vessels:
Vasoconstriction: When stimulated by norepinephrine, they cause the smooth muscle to contract. This:
Increases Systemic Vascular Resistance (SVR) and therefore increases Arterial Blood Pressure.
Constricts veins, which increases venous return of blood to the heart.
In the Heart:
A minor role in potentiating the effects of beta-receptor stimulation, leading to a slight increase in contractility.
2. Beta-Adrenoceptors (β-receptors)
Beta-receptors have diverse effects, primarily increasing heart function and causing vasodilation in certain beds.
Location and Function by Subtype:
A. Beta-1 Receptors (β₁)
Location: Primarily in the heart.
SA Node
AV Node
Atrial and Ventricular Myocardium
Function:
SA Node: Increase Heart Rate (positive chronotropy).
AV Node: Increase Conduction Velocity (positive dromotropy).
Myocardium: Increase Force of Contraction (positive inotropy).
Overall Effect: To significantly increase Cardiac Output.
B. Beta-2 Receptors (β₂)
Location: Primarily on vascular smooth muscle of specific vascular beds.
Skeletal Muscle Arterioles
Coronary Arteries (heart's own blood supply)
Arteries to the Liver
Function:
Vasodilation: When stimulated by epinephrine (and to a lesser extent, norepinephrine), they cause smooth muscle relaxation. This:
Decreases Systemic Vascular Resistance (SVR).
Redirects blood flow to skeletal muscle and the heart during "fight-or-flight."
Note: Beta-2 receptors are more sensitive to epinephrine (from the adrenal medulla) than to norepinephrine.
3. Muscarinic Receptors (M₂)
These are the primary receptors for parasympathetic (vagal) control of the heart. Parasympathetic innervation of most blood vessels is sparse, so the main cardiovascular effects are on the heart.
Location:
Heart: Primarily M₂ subtype.
SA Node
AV Node
Atrial Myocardium
(Ventricles have very few muscarinic receptors)
Blood Vessels:
Vascular Endothelium (inner lining): Muscarinic receptors (M₃) are present on the endothelial cells. They are not directly innervated by parasympathetic nerves but can be stimulated by circulating agonists.
Function:
In the Heart:
SA Node: Decreases Heart Rate (negative chronotropy) – the primary vagal effect.
AV Node: Decreases Conduction Velocity (negative dromotropy), which can slow or block transmission from atria to ventricles.
Atrial Myocardium: Decreases Force of Contraction (negative inotropy).
In Blood Vessels:
Indirect Vasodilation: When an agonist like ACh binds to M₃ receptors on the endothelium, it stimulates the production of Nitric Oxide (NO). NO diffuses to the underlying smooth muscle and causes relaxation and vasodilation.
Important Note: If the endothelium is damaged, ACh can bind to muscarinic receptors on the smooth muscle itself and cause weak vasoconstriction. This is a key difference in function based on location.
Summary Table for Quick Reference
Receptor Type | Primary Location | Effect when Stimulated | Mediator |
|---|---|---|---|
Alpha (α₁) | Vascular Smooth Muscle | Vasoconstriction (↑ BP) | Norepinephrine |
Beta-1 (β₁) | Heart (SA node, Myocardium) | ↑ HR, ↑ Contractility (↑ CO) | Norepinephrine |
Beta-2 (β₂) | Vascular Smooth Muscle (Skeletal, Coronary) | Vasodilation (↓ SVR) | Epinephrine |
Muscarinic (M₂) | Heart (SA node, AV node) | ↓ HR, ↓ Conduction Velocity | Acetylcholine (Vagus) |
Muscarinic (M₃) | Vascular Endothelium | Vasodilation (via NO) | Circulating Agonists |
Abbreviations: HR (Heart Rate), CO (Cardiac Output), BP (Blood Pressure), SVR (Systemic Vascular Resistance), NO (Nitric Oxide).
neurotransmitter and receptor for sympathetic nervous system
neurotransmitter and receptor for parasympathetic nervous system
Overview of Neurotransmitters
Sympathetic Nerves release Norepinephrine (and epinephrine from the adrenal medulla), which acts on alpha and beta-adrenoceptors.
Parasympathetic Nerves release Acetylcholine (ACh), which acts on muscarinic receptors.
summary table
Summary Table for Quick Reference
a2 is NOT IN THE CHART
Receptor Type | Primary Location | Effect when Stimulated | Mediator |
|---|---|---|---|
Alpha (α₁) (sympathetic) | Vascular Smooth Muscle | Vasoconstriction (↑ BP) | Norepinephrine |
Beta-1 (β₁) (sympathetic) | Heart (SA node, Myocardium) | ↑ HR, ↑ Contractility (↑ CO) | Norepinephrine |
Beta-2 (β₂) (sympathetic) | Vascular Smooth Muscle (Skeletal, Coronary) | Vasodilation (↓ SVR) | Epinephrine |
Muscarinic (M₂) (parasympathetic) | Heart (SA node, AV node) | ↓ HR, ↓ Conduction Velocity | Acetylcholine (Vagus) |
Muscarinic (M₃) (parasympathetic) | Vascular Endothelium | Vasodilation (via NO) | Circulating Agonists |
Abbreviations: HR (Heart Rate), CO (Cardiac Output), BP (Blood Pressure), SVR (Systemic Vascular Resistance), NO (Nitric Oxide).
a1 receptor (sympathetic nervous system)
a1 used to increase blood pressure
Alpha (α₁) | Vascular Smooth Muscle | Vasoconstriction (↑ BP) | Norepinephrine |
b1 receptor (sympathetic nervous system)
b1 used into increase cardiac ouput.
Beta-1 (β₁) | Heart (SA node, Myocardium) | ↑ HR, ↑ Contractility (↑ CO) | Norepinephrine |
b2 receptor (sympathetic nervous system)
b2 used to increase SVR
Beta-2 (β₂) | Vascular Smooth Muscle (Skeletal, Coronary) | Vasodilation (↓ SVR) | Epinephrine |
a1 and b2 are both located in vascular smooth muscle, but have opposing effects and different neurotransmitters.
what is the similarity and difference between a1 and b2 receptors?
a1 and b2 are both located in vascular smooth muscle, but have opposing effects and different neurotransmitters.
M2 receptor (parasympathetic nervous system)
Muscarinic (M₂) | Heart (SA node, AV node) | ↓ HR, ↓ Conduction Velocity | Acetylcholine (Vagus) |
M3 (parasympathetic nervous system)
Muscarinic (M₃) | Vascular Endothelium | Vasodilation (via NO) | Circulating Agonists |
what is the difference between m2 and m3 receptors?
location, m2 is heart, m3 is vascular endothelium
m2 decreases heart rate and conduction velocity through acetylcholine reception
m3 causes vasodilation via nitric oxide.
Describe the effects of sympathetic and parasympathetic stimulation on the heart and circulation.
The sympathetic and parasympathetic (vagal) divisions of the autonomic nervous system have opposing and complementary effects on the cardiovascular system, allowing for precise control of cardiac output and blood pressure to meet the body's changing demands.
Here is a detailed description of their effects.
Executive Summary: Core Effects
Sympathetic Stimulation ("Fight-or-Flight"): Increases Cardiac Output and Redirects Blood Flow. It prepares the body for action.
Parasympathetic Stimulation ("Rest-and-Digest"): Decreases Cardiac Output and Promotes Digestion. It conserves energy and maintains basal bodily functions.
1. Effects of Sympathetic Stimulation
Sympathetic activation is a system-wide response, primarily mediated by the release of norepinephrine from nerve endings and epinephrine from the adrenal medulla, acting on alpha and beta-adrenoceptors.
On the Heart:
The goal is to dramatically increase Cardiac Output (CO = Heart Rate x Stroke Volume).
Heart Rate (Chronotropy):
Effect: Marked Increase.
Mechanism: Stimulation of Beta-1 (β₁) receptors on the Sinoatrial (SA) node increases its rate of spontaneous depolarization, accelerating the heart rate (tachycardia).
Contractility (Inotropy):
Effect: Marked Increase.
Mechanism: Stimulation of Beta-1 (β₁) receptors on the atrial and ventricular myocardium enhances the force of contraction. This results in a more powerful squeeze, ejecting more blood with each beat and increasing Stroke Volume.
Conduction Velocity (Dromotropy):
Effect: Increase.
Mechanism: Stimulation of Beta-1 (β₁) receptors on the Atrioventricular (AV) node increases the speed of electrical impulse conduction from the atria to the ventricles. This ensures the rapidly beating chambers remain synchronized.
Net Cardiac Effect: A large increase in Cardiac Output, providing more oxygenated blood to the body.
On the Circulation (Blood Vessels):
The goal is to increase Blood Pressure and redirect blood flow from non-essential organs to active skeletal muscle and the heart.
Systemic Vascular Resistance (Afterload):
Effect: Variable, but net increase.
Mechanism:
Vasoconstriction: Strong stimulation of Alpha-1 (α₁) receptors on vascular smooth muscle in most beds (e.g., skin, gastrointestinal tract, kidneys) causes pronounced vasoconstriction. This dramatically increases systemic vascular resistance (SVR) and arterial blood pressure.
Vasodilation: Stimulation of Beta-2 (β₂) receptors on vascular smooth muscle in skeletal muscle and the liver causes vasodilation. This shunts blood away from constricted areas and toward the muscles needed for action.
Venous Return (Preload):
Effect: Increase.
Mechanism: Constriction of veins (via α₁-receptors) reduces their capacity to hold blood. This "squeezes" more blood back to the heart, increasing venous return and preload, which further enhances stroke volume via the Frank-Starling mechanism.
Net Circulatory Effect: Elevated blood pressure and a redistribution of blood flow, prioritizing the brain, heart, and working muscles.
2. Effects of Parasympathetic (Vagal) Stimulation
Parasympathetic effects are more localized and discrete, mediated almost exclusively by the release of acetylcholine from the vagus nerve (CN X), acting on muscarinic receptors.
On the Heart:
The goal is to decrease Cardiac Output and conserve energy.
Heart Rate (Chronotropy):
Effect: Marked Decrease.
Mechanism: Stimulation of Muscarinic (M₂) receptors on the SA node hyperpolarizes the cells and decreases their rate of depolarization, significantly slowing the heart rate (bradycardia). This is the most powerful and pronounced effect of vagal stimulation.
Contractility (Inotropy):
Effect: Mild to Moderate Decrease (in atria).
Mechanism: Stimulation of M₂ receptors in the atrial myocardium reduces the force of contraction. The ventricles receive very little parasympathetic innervation, so the effect on overall contractility is limited.
Conduction Velocity (Dromotropy):
Effect: Decrease.
Mechanism: Stimulation of M₂ receptors on the AV node slows down impulse conduction, increasing the PR interval on an ECG. High vagal tone can even lead to a transient AV block.
Net Cardiac Effect: A pronounced decrease in Heart Rate, leading to a significant reduction in Cardiac Output.
On the Circulation (Blood Vessels):
Systemic Vascular Resistance:
Effect: Minimal Direct Effect.
Mechanism: Most blood vessels do not receive parasympathetic innervation. Therefore, vagal stimulation has little direct control over systemic vascular resistance. The primary control of vasodilation/vasoconstriction is sympathetic.
Exception: Parasympathetic nerves do cause vasodilation in specific, localized beds such as the salivary glands, glands of the GI tract, and external genitalia (erection) to support "rest-and-digest" functions. This is not a system-wide effect.
Summary Table: Opposing Effects
Target Organ | Sympathetic Effect | Parasympathetic Effect |
|---|---|---|
SA Node | ↑↑ Heart Rate (Tachycardia) | ↓↓ Heart Rate (Bradycardia) |
AV Node | ↑ Conduction Velocity | ↓ Conduction Velocity (AV Block) |
Atrial Muscle | ↑ Contractility | ↓ Contractility |
Ventricular Muscle | ↑↑ Contractility | Very Little Effect |
Arterioles | Constriction (most beds) / Dilation (skeletal muscle) | No Innervation (most beds) |
Veins | Constriction (↑ Venous Return) | No Innervation |
Overall Result | ↑ Cardiac Output, ↑ BP, Redirect Flow | ↓↓ Cardiac Output, Conserve Energy |
Physiological Integration
In a healthy person, there is a constant, dynamic interplay between these two systems—a phenomenon known as "autonomic tone."
At rest, parasympathetic (vagal) tone is dominant, which keeps the heart rate lower than the intrinsic rate of the SA node (e.g., 60-80 bpm instead of 100 bpm).
During exercise or stress, sympathetic tone increases and parasympathetic tone withdraws, allowing for a rapid and powerful increase in heart rate and contractility.
This precise balance allows for the fine-tuned, moment-to-moment regulation required to maintain cardiovascular homeostasis.
List the anatomical components of the baroreceptor reflex.
The baroreceptor reflex is the body's primary short-term mechanism for maintaining stable blood pressure. Its anatomical components form a classic negative feedback loop, comprising sensors, afferent pathways, an integration center, efferent pathways, and effector organs.
Here is a list of the key anatomical components.
Anatomical Components of the Baroreceptor Reflex1. Sensors (Baroreceptors / Pressoreceptors)
Location: Specialized stretch receptors located in the walls of specific arteries.
Carotid Sinus: A slight dilation at the bifurcation of the common carotid artery, just before it branches into the internal and external carotid arteries. This is the most important site.
Aortic Arch: Located in the wall of the arch of the aorta.
Function: These receptors are sensitive to changes in arterial pressure. An increase in BP causes increased stretch of the arterial wall, leading to increased baroreceptor firing. A decrease in BP causes less stretch and decreased firing.
2. Afferent (Sensory) Pathways
Nerves: These carry the sensory information from the baroreceptors to the brain.
Glossopharyngeal Nerve (Cranial Nerve IX): Carries signals from the carotid sinus baroreceptors.
Vagus Nerve (Cranial Nerve X): Carries signals from the aortic arch baroreceptors.
3. Integration Center
Location: The Cardiovascular Center in the Medulla Oblongata of the brainstem.
Key Components:
Nucleus of the Solitary Tract (NTS): This is the primary site where the afferent nerves (CN IX and X) first synapse and relay the sensory input.
Other Medullary Centers: The NTS communicates with adjacent centers to coordinate the response:
Cardioinhibitory Center: Promotes parasympathetic (vagal) output.
Vasomotor Center: Controls sympathetic output to the heart and blood vessels.
4. Efferent (Motor) Pathways
Parasympathetic Pathway:
Nerve: The Vagus Nerve (Cranial Nerve X).
Target: The heart (SA node, AV node, atria).
Sympathetic Pathway:
Path: Preganglionic neurons from the vasomotor center descend the spinal cord and synapse in the intermediolateral (IML) cell column (T1-L2). Postganglionic neurons then exit the cord.
Nerves: Cardiac and Vasomotor Sympathetic Nerves.
Targets: The heart (SA node, AV node, myocardium) and vascular smooth muscle in arterioles and veins.
5. Effector Organs
The Heart:
SA Node: Changes heart rate.
AV Node: Changes conduction velocity.
Cardiac Muscle (Myocardium): Changes contractility (force of contraction).
Blood Vessels:
Arterioles (Resistance Vessels): Changes vascular tone, altering systemic vascular resistance (SVR).
Veins (Capacitance Vessels): Changes venous tone, altering venous return and preload.
Summary of the Reflex Arc in Action
Component | Anatomy Involved |
|---|---|
Sensor | Baroreceptors in Carotid Sinus & Aortic Arch |
Afferent Pathway | Glossopharyngeal Nerve (IX) & Vagus Nerve (X) |
Integration Center | Medulla Oblongata (NTS, Cardioinhibitory & Vasomotor Centers) |
Efferent Pathway | Vagus Nerve (X) & Sympathetic Nerves |
Effectors | Heart & Blood Vessels |
This elegant reflex allows for moment-to-moment adjustments, such as preventing a drop in blood pressure when you stand up (orthostatic hypotension) or moderating a spike in pressure during a stress response.
Describe how carotid sinus baroreceptors respond to changes in arterial pressure (mean pressure and pulse pressure), and explain how changes in baroreceptor activity affect sympathetic and parasympathetic outflow to the heart and circulation.
This is a core concept in cardiovascular physiology. Here is a detailed description of how carotid sinus baroreceptors function and how their signals are translated into autonomic responses.
1. How Baroreceptors Respond to Changes in Arterial Pressure
Carotid sinus baroreceptors are stretch-sensitive nerve endings embedded in the wall of the carotid sinus. They do not respond to pressure directly, but to the stretch of the arterial wall caused by that pressure.
Response to Mean Arterial Pressure (MAP)
MAP is the average pressure in the arteries throughout one cardiac cycle, representing the perfusion pressure driving blood to the tissues.
Relationship: Baroreceptor firing rate is directly proportional to MAP, but this relationship is not linear across all pressures. It follows a sigmoidal (S-shaped) curve.
Threshold: Below a MAP of ~50-60 mmHg, the vessel wall is under too little stretch, and baroreceptors are silent.
Sensitive Range: Between ~60-180 mmHg, the baroreceptors are exquisitely sensitive. Small changes in MAP produce large changes in firing rate. The normal operating point (around 100 mmHg) lies within this steep, sensitive region, allowing for precise control.
Saturation: Above ~180 mmHg, the vessel wall is maximally stretched, and baroreceptor firing reaches a plateau and cannot increase further.
Response to Pulse Pressure
Pulse Pressure is the difference between systolic and diastolic pressure (SBP - DBP). It is a reflection of stroke volume and arterial stiffness.
Relationship: At any given MAP, baroreceptors are more sensitive to a pulsatile pressure than to a steady pressure.
Mechanism: The rhythmic stretching and recoiling of the artery wall during each cardiac cycle cause a burst of baroreceptor action potentials with each pulse.
Effect: A higher pulse pressure (e.g., due to a larger stroke volume) results in a greater peak stretch during systole, leading to a higher overall average firing rate than a steady pressure at the same MAP would produce.
In summary: Baroreceptor firing encodes both the mean level of arterial pressure (over the sensitive range) and its pulsatile nature. Increased MAP or increased Pulse Pressure leads to increased baroreceptor firing.
2. How Baroreceptor Activity Affects Autonomic Outflow
The primary integration center for the baroreceptor reflex is the medulla oblongata, specifically the Nucleus of the Solitary Tract (NTS). The autonomic response is a classic negative feedback loop.
Scenario 1: Response to a RISE in Arterial Pressure (e.g., during stress)
Stimulus: ↑ Arterial Pressure → ↑ Stretch of Carotid Sinus.
Sensor: ↑ Firing rate of Carotid Sinus Baroreceptors.
Afferent Pathway: ↑ Firing in the Glossopharyngeal Nerve (CN IX).
Integration (Medulla):
The NTS is activated.
It stimulates the Cardioinhibitory Center (Parasympathetic).
It inhibits the Vasomotor Center (Sympathetic).
Efferent Outflow & Effector Response:
↑ Parasympathetic Outflow (Vagus Nerve):
Heart: Acts on the SA node to decrease Heart Rate (Bradycardia).
↓ Sympathetic Outflow:
Heart: Decreases heart rate and contractility.
Blood Vessels: Causes vasodilation in arterioles (decreasing Systemic Vascular Resistance) and veins (decreasing Venous Return).
Net Effect: ↓ Heart Rate, ↓ Contractility, ↓ Venous Return, ↓ Systemic Vascular Resistance → ↓ Cardiac Output & ↓ Arterial Pressure back toward normal.
Scenario 2: Response to a FALL in Arterial Pressure (e.g., upon standing)
Stimulus: ↓ Arterial Pressure → ↓ Stretch of Carotid Sinus.
Sensor: ↓ Firing rate of Carotid Sinus Baroreceptors.
Afferent Pathway: ↓ Firing in the Glossopharyngeal Nerve (CN IX).
Integration (Medulla):
The NTS is less activated.
It inhibits the Cardioinhibitory Center (Parasympathetic).
It stimulates the Vasomotor Center (Sympathetic).
Efferent Outflow & Effector Response:
↓ Parasympathetic Outflow (Vagus Nerve):
Heart: Withdraws the "brake" on the SA node, leading to increase Heart Rate (Tachycardia). This is often the fastest response.
↑ Sympathetic Outflow:
Heart: Increases heart rate and contractility (via β₁-receptors), boosting Stroke Volume and Cardiac Output.
Blood Vessels: Causes widespread vasoconstriction in arterioles (increasing Systemic Vascular Resistance) and veins (increasing Venous Return and Preload).
Net Effect: ↑ Heart Rate, ↑ Contractility, ↑ Venous Return, ↑ Systemic Vascular Resistance → ↑ Cardiac Output & ↑ Arterial Pressure back toward normal.
Summary Diagram of the Reflex Arc
Change in BP | Baroreceptor Firing | Parasympathetic Outflow | Sympathetic Outflow | Final Effect on BP |
|---|---|---|---|---|
↑ Pressure | Increases | Increases (↓ HR) | Decreases (Vasodilation, ↓ Contractility) | Decreases (Corrective) |
↓ Pressure | Decreases | Decreases (↑ HR) | Increases (Vasoconstriction, ↑ Contractility) | Increases (Corrective) |
This elegant reflex is essential for maintaining moment-to-moment stability of blood pressure, ensuring adequate perfusion to the brain and other vital organs during routine activities like changing posture.
Explain the sequence of events mediated by cardiopulmonary (volume) receptors that occur after an acute increase or decrease in arterial blood pressure.
This is an excellent question that highlights a crucial, complementary system to the arterial baroreceptor reflex.
While arterial baroreceptors (in the carotid sinus and aorta) are primarily sensitive to short-term pressure changes, the cardiopulmonary receptors (also called low-pressure or volume receptors) are primarily sensitive to changes in blood volume and central venous pressure.
Here is the sequence of events mediated by these receptors in response to acute changes in blood pressure, particularly those stemming from volume shifts.
Overview: Location and Stimulus of Cardiopulmonary Receptors
Location: These receptors are located in the walls of the atria (especially at the venoatrial junctions) and the pulmonary vasculature.
Stimulus: They are "low-pressure" receptors because they sense pressure (or, more accurately, stretch) in the cardiac chambers and pulmonary circulation, which is much lower than in the systemic arteries. This stretch is directly influenced by:
Blood Volume
Central Venous Pressure (CVP)
Venous Return
An acute change in arterial blood pressure often reflects an underlying change in volume or venous return, which is then detected by these cardiopulmonary receptors.
Scenario 1: After an ACUTE INCREASE in Arterial Pressure (e.g., due to rapid fluid infusion)
An increase in blood volume leads to an increase in venous return, central venous pressure, and ultimately, arterial pressure.
Sequence of Events:
Stimulus: Increased blood volume → Increased venous return → Increased Central Venous Pressure (CVP) → Increased stretch of the atrial walls.
Receptor Activation: Cardiopulmonary Receptors in the atria and pulmonary arteries are stimulated (increased firing rate).
Afferent Pathway: Signals are sent to the medulla via the Vagus Nerve (Cranial Nerve X).
Central Integration (in the Medulla):
The Nucleus of the Solitary Tract (NTS) is activated.
This leads to two major responses:
Inhibition of the Vasomotor Center: Decreasing sympathetic outflow.
Stimulation of the Cardioinhibitory Center: Increasing parasympathetic (vagal) outflow.
Efferent and Effector Responses (The "3 Ds"):
Decreased Sympathetic Outflow:
To the Kidneys: Leads to vasodilation of renal arterioles. This, combined with the direct inhibition of sympathetic tone to the juxtaglomerular (JG) cells, causes a marked decrease in Renin release. This suppresses the Renin-Angiotensin-Aldosterone System (RAAS), reducing sodium and water reabsorption.
To the Systemic Arterioles: Causes vasodilation, helping to lower systemic vascular resistance and arterial pressure.
To the Veins: Reduces venous tone.
Increased Parasympathetic Outflow:
To the SA Node: Decreases heart rate (a minor effect compared to the renal effects).
Hormonal Response:
The increased atrial stretch directly causes the atria to secrete Atrial Natriuretic Peptide (ANP). ANP promotes vasodilation and, crucially, increases sodium and water excretion by the kidneys (natriuresis and diuresis).
Net Result: A coordinated effort to reduce blood volume and pressure through:
Vasodilation (decreasing peripheral resistance).
Increased water and salt excretion by the kidneys (decreasing blood volume).
A slight decrease in heart rate.
Scenario 2: After an ACUTE DECREASE in Arterial Pressure (e.g., due to hemorrhage)
A decrease in blood volume leads to decreased venous return, central venous pressure, and arterial pressure.
Sequence of Events:
Stimulus: Decreased blood volume → Decreased venous return → Decreased Central Venous Pressure (CVP) → Decreased stretch of the atrial walls.
Receptor Activation: Cardiopulmonary Receptors are unloaded (decreased firing rate).
Afferent Pathway: Decreased signaling to the medulla via the Vagus Nerve.
Central Integration (in the Medulla):
The Nucleus of the Solitary Tract (NTS) is less activated.
This leads to:
Stimulation of the Vasomotor Center: Increasing sympathetic outflow.
Inhibition of the Cardioinhibitory Center: Decreasing parasympathetic (vagal) outflow.
Efferent and Effector Responses (The "3 Cs" - Conservation and Constriction):
Increased Sympathetic Outflow:
To the Kidneys: Causes potent vasoconstriction of renal arterioles and directly stimulates Renin release from the JG cells. This activates the RAAS, leading to angiotensin II-mediated vasoconstriction and aldosterone-mediated sodium and water retention.
To the Systemic Arterioles: Causes widespread vasoconstriction, increasing systemic vascular resistance to maintain arterial pressure.
To the Veins: Increases venous tone, which helps maintain venous return.
Decreased Parasympathetic Outflow:
To the SA Node: Increases heart rate (Tachycardia) to help maintain cardiac output.
Hormonal Response:
Decreased atrial stretch leads to a decrease in ANP release, removing its inhibitory effects on sodium reabsorption and vasodilation.
Net Result: A coordinated effort to conserve blood volume and maintain pressure through:
Vasoconstriction (increasing peripheral resistance).
Conservation of water and salt by the kidneys (conserving blood volume).
An increase in heart rate.
Summary Table
Event | After ACUTE INCREASE in Pressure/Volume | After ACUTE DECREASE in Pressure/Volume |
|---|---|---|
Stimulus | ↑ CVP, ↑ Atrial Stretch | ↓ CVP, ↓ Atrial Stretch |
Receptor Firing | Increases | Decreases |
Sympathetic Outflow | Marked Decrease | Marked Increase |
Parasympathetic Outflow | Increase | Decrease |
Renal Sympathetic Nerve Activity | Decreased → ↓ Renin, ↑ Renal Blood Flow | Increased → ↑ Renin, ↓ Renal Blood Flow |
Key Hormone | ↑ ANP (causes Na+/water excretion) | ↑ Aldosterone (causes Na+/water retention) |
Overall Goal | Reduce Blood Volume & Pressure | Conserve Blood Volume & Maintain Pressure |
In conclusion, the cardiopulmonary reflex works in tandem with the arterial baroreflex. While the arterial reflex is the first line of defense against pressure fluctuations, the cardiopulmonary reflex is crucial for the long-term regulation of blood volume, primarily by controlling sympathetic outflow to the kidneys and the release of key hormones.
Describe (a) the location of peripheral and central chemoreceptors; (b) the way they respond to hypoxemia, hypercapnia, and acidosis, and 9c) the effects of their stimulation in autonomic control of the heart and circulation.
Here is a detailed description of the peripheral and central chemoreceptors, their responses, and their cardiovascular effects.
Overview
Chemoreceptors are specialized sensors that monitor the chemical composition of the blood and cerebrospinal fluid (CSF). Their primary role is to regulate ventilation, but their stimulation also has significant effects on the autonomic control of the cardiovascular system. The two main groups are the peripheral and central chemoreceptors.
(a) Location of Chemoreceptors
Chemoreceptor Type | Specific Name & Location |
|---|---|
Peripheral Chemoreceptors | Located outside the blood-brain barrier. |
Carotid Bodies: Bifurcation of the common carotid arteries. (Most important for cardiovascular responses). | |
Aortic Bodies: Between the arch of the aorta and pulmonary artery. | |
Central Chemoreceptors | Located on the ventrolateral surface of the medulla oblongata, bathed in the cerebrospinal fluid (CSF). |
(b) Response to Stimuli (Hypoxemia, Hypercapnia, Acidosis)
The peripheral and central chemoreceptors have different sensitivities to these chemical stimuli.
Peripheral Chemoreceptors (Carotid and Aortic Bodies)
Hypoxemia (Low PaO₂):
Response: Extremely sensitive. They are the primary sensors for low oxygen in the arterial blood. Their firing rate increases exponentially once the arterial PaO₂ falls below ~60 mmHg.
Hypercapnia (High PaCO₂):
Response: Sensitive. They respond directly to an increase in arterial CO₂. However, their response is rapid but less potent than the central chemoreceptors over time.
Acidemia (Low arterial pH / Metabolic Acidosis):
Response: Very sensitive. They respond directly to a decrease in arterial blood pH. This is a powerful stimulus.
Key Point: The effects of hypercapnia and acidemia are synergistic. A combined high CO₂ and low pH (as in respiratory acidosis) produces a much stronger response than either alone.
Central Chemoreceptors (Medulla)
Hypoxemia (Low PaO₂):
Response: Not directly stimulated. They are not sensitive to low oxygen per se. However, severe hypoxemia (PaO₂ < 50 mmHg) can depress neuronal function, including the central chemoreceptors.
Hypercapnia (High PaCO₂):
Response: Exquisitely sensitive. This is their primary stimulus. CO₂ readily diffuses across the blood-brain barrier into the CSF.
Mechanism: Inside the CSF, CO₂ combines with water to form carbonic acid (H₂CO₃), which dissociates into H⁺ and bicarbonate (HCO₃⁻). It is the increase in H⁺ ion concentration (a drop in CSF pH) that directly stimulates the central chemoreceptors.
Acidemia (Low arterial pH / Metabolic Acidosis):
Response: Poorly responsive. The blood-brain barrier is relatively impermeable to H⁺ ions. Therefore, a metabolic acidosis in the blood has a slow and limited effect on the central chemoreceptors compared to a respiratory acidosis (caused by high CO₂).
(c) Effects of Stimulation on Autonomic Control of Heart and Circulation
When chemoreceptors are stimulated, they trigger a complex response that involves both the respiratory and cardiovascular centers in the medulla. The final cardiovascular outcome depends on the interplay between a primary reflex and a secondary override from lung inflation.
1. The Primary Chemoreceptor Reflex (Apneic Period)
If the chemoreceptors are stimulated while holding one's breath (e.g., during a dive), the primary, unmodified reflex is seen:
Afferent Signal: Chemoreceptor stimulation sends signals via the glossopharyngeal (IX) and vagus (X) nerves to the medullary cardiovascular centers.
Efferent Response:
Massive Increase in Sympathetic Outflow: This causes intense, widespread vasoconstriction in skeletal muscle, renal, and splanchnic circulations. The goal is to drastically increase systemic vascular resistance (SVR) to maintain blood pressure and redirect blood flow to the heart and brain.
Parasympathetic (Vagal) Outflow to the Heart: This causes profound bradycardia (a very low heart rate).
Net Effect: Bradycardia + Hypertension.
Physiological Example: This is the diving reflex, an oxygen-conserving response.
2. The Integrated Chemoreceptor Reflex (During Spontaneous Breathing)
Under normal conditions, chemoreceptor stimulation (e.g., from hypoxia) simultaneously triggers hyperventilation. The lung inflation associated with breathing initiates a secondary reflex that modifies the primary response.
Step 1: Primary Reflex Occurs: As above, there is an initial tendency toward bradycardia and vasoconstriction.
Step 2: The Override from Lung Stretch Receptors:
Inflating the lungs stimulates pulmonary stretch receptors.
These send signals that inhibit cardiac vagal tone (reducing bradycardia) and inhibit sympathetic vasoconstrictor outflow.
The Final, Observed Cardiovascular Response:
Heart Rate: The bradycardia is typically overridden, leading to a mild tachycardia (increased heart rate) due to the withdrawal of vagal tone.
Vascular Tone: The widespread sympathetic vasoconstriction is modulated. It is still present but becomes more regionalized. Vasoconstriction is maintained in non-essential beds (e.g., gut, kidney), but there can be sympathetically-mediated vasodilation in skeletal muscle (via β₂-adrenoceptors) to facilitate oxygen delivery.
Cardiac Output: Increases due to tachycardia.
Blood Pressure: Usually rises modestly due to increased cardiac output and regional vasoconstriction.
Summary Table of Effects
Scenario | Heart Rate | Systemic Vascular Resistance | Blood Pressure | Key Driver |
|---|---|---|---|---|
Primary Reflex (No breathing) | ↓↓ (Bradycardia) | ↑↑ (Massive Vasoconstriction) | ↑↑ (Hypertension) | Chemoreceptor input alone |
Integrated Reflex (With breathing) | ↑ (Tachycardia) | ↑ (Regional Vasoconstriction) | ↑ (Mild Hypertension) | Lung inflation overrides vagal bradycardia |
In summary, chemoreceptors are crucial for detecting blood gas abnormalities. While their primary job is to adjust breathing, their stimulation triggers powerful autonomic cardiovascular adjustments aimed at preserving oxygen delivery to vital organs, with the final outcome being finely tuned by concurrent respiratory activity.
List the factors that stimulate the release of catecholamines, renin, atrial natriuretic peptide, and vasopressin.
Here is a detailed list of the factors that stimulate the release of these key cardiovascular hormones.
1. Catecholamines (Epinephrine and Norepinephrine)
Primary Source: Adrenal medulla (mainly epinephrine), and sympathetic nerve endings (norepinephrine).
Stimulus Category | Specific Factors |
|---|---|
Neural (Primary) | ✓ Direct Sympathetic Stimulation: Preganglionic sympathetic fibers (via acetylcholine and nicotinic receptors) stimulate the adrenal medulla to secrete catecholamines directly into the bloodstream. This is the fastest mechanism. |
Stress & Homeostatic Threats | ✓ Hypovolemia / Hemorrhage: Detected by baroreceptors and volume receptors. |
Other Hormones | ✓ Angiotensin II: Can potentiate the release of catecholamines from the adrenal medulla. |
2. Renin
Primary Source: Juxtaglomerular (JG) cells in the afferent arterioles of the kidney.
Stimulus Category | Specific Factors |
|---|---|
Renal Perfusion Pressure | ✓ Reduced Renal Perfusion Pressure: A drop in systemic BP (e.g., hemorrhage) or renal artery stenosis decreases stretch of the afferent arteriole, directly stimulating the Baroreceptor Mechanism in the JG cells. |
Sympathetic Nervous System | ✓ Increased Renal Sympathetic Nerve Activity: Direct stimulation of β₁-adrenoceptors on the JG cells. This occurs during stress, exercise, or heart failure. |
Tubular Composition | ✓ Decreased Sodium Delivery to the Macula Densa: A low NaCl load in the distal tubule (due to low GFR or high proximal reabsorption) is detected by the macula densa, which signals the JG cells to release renin via the Macula Densa Mechanism. |
3. Atrial Natriuretic Peptide (ANP)
Primary Source: Cardiac atrial myocytes.
Stimulus Category | Specific Factors |
|---|---|
Cardiac Wall Stretch (Primary) | ✓ Increased Atrial Stretch / Pressure: This is the major stimulus. It occurs during: |
Neurohormonal Factors | ✓ Endothelin & α-adrenergic agonists: Can stimulate ANP release, though the stretch mechanism is dominant. |
4. Vasopressin (Antidiuretic Hormone - ADH)
Primary Source: Supraoptic and paraventricular nuclei of the hypothalamus, released from the posterior pituitary.
Stimulus Category | Specific Factors |
|---|---|
Osmoregulation (Primary) | ✓ Increased Plasma Osmolality: A 1-2% increase, primarily due to elevated sodium concentration, is detected by osmoreceptors in the hypothalamus. This is the most sensitive stimulus for everyday regulation. |
Volume/Pressure Regulation | ✓ Significant Hypovolemia / Hemorrhage: A >5-10% drop in blood volume is detected by cardiopulmonary and arterial baroreceptors. This unloads their inhibitory input to the hypothalamus, leading to a powerful release of ADH. |
Other Stimuli | ✓ Angiotensin II: Acts on the hypothalamus to stimulate thirst and ADH release. |
Summary Table for Quick Reference
Hormone | Primary Stimuli for Release |
|---|---|
Catecholamines | Sympathetic activation (stress, hypotension, hypoglycemia, hypoxemia) |
Renin | 1. Low Renal Perfusion Pressure |
Atrial Natriuretic Peptide (ANP) | Increased Atrial Stretch/Pressure (hypervolemia, heart failure) |
Vasopressin (ADH) | 1. High Plasma Osmolality |
This coordinated hormonal response is crucial for maintaining blood pressure, blood volume, and electrolyte balance during various physiological challenges.
Describe how the sympathetic nerves, circulating catecholamines, angiotensin II, aldosterone, atrial natriuretic peptide, and vasopressin interact to regulate arterial blood pressure.
The regulation of arterial blood pressure is a complex interplay of neural and hormonal systems that act on two primary variables: Cardiac Output (CO) and Systemic Vascular Resistance (SVR), according to the equation BP = CO × SVR.
These systems interact in a coordinated, often hierarchical, network to maintain homeostasis. The following chart illustrates these interactions, showing how different systems work together to raise (pressor) or lower (depressor) blood pressure:
Detailed Explanation of Interactions
Here is a detailed breakdown of how each factor works and interacts, as shown in the chart:
1. The Pressor Systems: Responding to Low BP/Volume
These systems are activated by a drop in blood pressure or blood volume (e.g., hemorrhage, dehydration).
A. Sympathetic Nervous System (SNS) & Catecholamines
Direct Action:
Heart: Increases heart rate and contractility (↑ Cardiac Output).
Vessels: Causes vasoconstriction (↑ Systemic Vascular Resistance).
Interaction with Others:
Stimulates RAAS: Directly triggers renin release from the kidneys.
Synergizes with Angiotensin II: Both are potent vasoconstrictors; their effects are additive.
Stimulates Vasopressin Release: SNS activation can contribute to ADH release.
B. The Renin-Angiotensin-Aldosterone System (RAAS)
Direct Action:
Angiotensin II: A very potent vasoconstrictor (↑ SVR). It also stimulates thirst.
Aldosterone: Promotes sodium (and thus water) reabsorption in the kidneys, increasing blood volume (↑ Preload → ↑ Stroke Volume → ↑ CO).
Interaction with Others:
Stimulated by SNS & Low Renal Pressure.
Angiotensin II Potentiates SNS: Increases norepinephrine release from nerves and promotes central SNS outflow.
Angiotensin II Stimulates Vasopressin Release.
C. Vasopressin (Antidiuretic Hormone, ADH)
Direct Action:
V2 Receptors (Kidneys): Promotes water reabsorption (water retention), increasing blood volume.
V1 Receptors (Vessels): Causes vasoconstriction (↑ SVR), especially at high concentrations.
Interaction with Others:
Stimulated by Angiotensin II and SNS.
Works in parallel with Aldosterone to increase blood volume, but via water retention rather than sodium retention.
Summary of Pressor Interactions: A fall in BP triggers a coordinated response where the SNS, RAAS, and Vasopressin work synergistically to increase heart function, vasoconstriction, and blood volume, thereby raising blood pressure.
2. The Depressor System: Responding to High BP/Volume
This system counteracts the pressor systems to prevent hypertension and volume overload.
D. Atrial Natriuretic Peptide (ANP)
Direct Action:
Kidneys: Promotes excretion of sodium and water (natriuresis and diuresis), reducing blood volume (↓ CO).
Vessels: Causes vasodilation (↓ SVR).
Interaction with Others (Inhibitory):
Inhibits Renin Release: Suppresses the RAAS axis at its start.
Inhibits Aldosterone Release: Directly and indirectly via renin inhibition.
Inhibits Vasopressin Release.
Reduces Sympathetic Outflow: Centrally and peripherally.
Antagonizes Angiotensin II: Counteracts its vasoconstrictive effects.
Integrated Scenario Example
In Heart Failure, the system becomes maladaptive:
Low cardiac output is perceived as "low volume."
This chronically activates the Pressor Systems (SNS, RAAS, Vasopressin).
While this initially helps support BP, it leads to excessive fluid retention (worsening edema) and increased afterload (vasoconstriction), further straining the heart.
ANP levels are high but are often overwhelmed, a state known as "ANP resistance."
Treatment involves using drugs (e.g., ACE inhibitors, beta-blockers) to inhibit the overactive pressor systems and rebalance the equation.
In conclusion, arterial blood pressure is not regulated by a single hormone but by a dynamic, interconnected network where pressor and depressor forces constantly interact to maintain stability.
Describe the autonomic and hormonal compensatory mechanisms that are activated to restore arterial pressure following hemorrhage.
The compensatory mechanisms following hemorrhage are a classic and excellent example of the body's integrated physiological response to a life-threatening challenge. The goal is to restore arterial pressure and, ultimately, tissue perfusion.
These mechanisms are activated in a time-dependent sequence, ranging from immediate neural reflexes to longer-term hormonal and renal adjustments.
Overview of the Challenge
Hemorrhage → Loss of blood volume → ↓ Venous return → ↓ Stroke Volume → ↓ Cardiac Output → ↓ Arterial Blood Pressure.
↓ Arterial Pressure → ↓ Perfusion of vital organs (brain, heart, kidneys).
The body's response is to increase Cardiac Output (CO) and Systemic Vascular Resistance (SVR) to restore blood pressure: BP = CO x SVR.
Compensatory Mechanisms
The following flowchart illustrates the sequence and interaction of these compensatory mechanisms following a hemorrhage:
Detailed Breakdown of Mechanisms1. Immediate Compensatory Mechanisms (Within Seconds to Minutes)
These are primarily neural and are the body's first and fastest line of defense.
Baroreceptor Reflex:
Stimulus: ↓ Arterial Pressure → decreased stretch of carotid sinus and aortic arch baroreceptors.
Response:
↓ Parasympathetic (Vagal) Outflow: Rapid withdrawal of vagal tone to the heart leads to an immediate increase in heart rate (tachycardia).
↑ Sympathetic Outflow: This is the cornerstone of the early response.
Heart: Increases heart rate and contractility, attempting to boost Cardiac Output.
Arterioles: Causes widespread vasoconstriction (especially in skin, splanchnic organs, and kidneys). This dramatically increases Systemic Vascular Resistance (SVR), shunting blood away from non-vital organs to preserve flow to the heart and brain.
Veins: Causes venoconstriction, which decreases venous capacitance and "mobilizes" stored blood, increasing venous return.
Chemoreceptor Reflex:
Stimulus: If hypotension is severe enough to reduce O₂ delivery, hypoxemia and acidosis stimulate peripheral chemoreceptors.
Response: Further amplifies sympathetic vasoconstriction and contributes to the sensation of air hunger (increased ventilation).
2. Intermediate Compensatory Mechanisms (Within Minutes to Hours)
These hormonal systems reinforce the sympathetic response and begin to address the root problem: the loss of blood volume.
Activation of the Renin-Angiotensin-Aldosterone System (RAAS):
Stimulus: ↓ Renal perfusion pressure, renal sympathetic stimulation (β₁-adrenergic), and decreased sodium delivery to the macula densa.
Response:
Renin is released by the kidneys, leading to the formation of Angiotensin II.
Angiotensin II:
Potent vasoconstriction (↑ SVR), even more powerful than norepinephrine.
Stimulates the release of Aldosterone from the adrenal cortex and Vasopressin from the pituitary.
Stimulates thirst.
Aldosterone: Acts on the kidneys to promote sodium and water reabsorption. This is crucial for restoring blood volume.
Release of Vasopressin (Antidiuretic Hormone, ADH):
Stimulus: ↓ Blood volume (via cardiopulmonary receptors), ↓ BP (via baroreceptors), and Angiotensin II.
Response:
V1 Receptors: Causes vasoconstriction (↑ SVR).
V2 Receptors: Promotes water reabsorption in the kidneys, conserving body water and helping to restore plasma volume.
Circulating Catecholamines:
Stimulus: Sympathetic stimulation of the adrenal medulla.
Response: Epinephrine and some norepinephrine are released into the bloodstream. They reinforce the direct sympathetic effects by increasing heart rate, contractility, and vasoconstriction.
3. Long-Term Compensatory Mechanisms (Hours to Days)
Fluid Shifts (Transcapillary Reabsorption):
Mechanism: The drop in capillary hydrostatic pressure caused by hypotension and vasoconstriction disrupts Starling's forces. This favors the movement of fluid from the interstitial space into the capillaries.
Effect: Hemodilution (decreased hematocrit) and an increase in plasma volume. This is a purely physical process but is critical for volume restoration.
Increased Thirst and Sodium Appetite:
Stimulus: Angiotensin II and other signals act on the hypothalamus.
Effect: Drives behavioral changes to increase fluid and salt intake, correcting the volume deficit.
Renal Conservation of Water and Salt:
Mechanism: Sustained action of Aldosterone and Vasopressin over hours to days ensures that the kidneys retain as much water and sodium as possible, minimizing losses.
Summary of the Integrated Response
Target | Compensatory Change | Primary Mediator(s) | Effect on BP |
|---|---|---|---|
Heart | ↑ Heart Rate, ↑ Contractility | SNS, Catecholamines | ↑ Cardiac Output |
Arterioles | Widespread Vasoconstriction | SNS, Angiotensin II, Vasopressin | ↑ Systemic Vascular Resistance |
Veins | Venoconstriction | SNS | ↑ Venous Return |
Kidneys | Na⁺ and Water Retention | Aldosterone, Vasopressin, RAAS | ↑ Blood Volume → ↑ Cardiac Output |
Fluid Compartments | Transcapillary Reabsorption | Physical forces (↓ Capillary Pressure) | ↑ Plasma Volume |
This multi-level, redundant system is highly effective at compensating for moderate blood loss. However, with severe or prolonged hemorrhage, these mechanisms can become overwhelmed, leading to a vicious cycle of decompensation and circulatory shock.
List the mechanisms and causes of hypotension.
Hypotension, or low blood pressure, can result from a failure in any of the body's mechanisms that maintain arterial pressure. These can be broadly categorized by the underlying problem: a decrease in cardiac output (CO) or a decrease in systemic vascular resistance (SVR), according to the equation BP = CO x SVR.
Here is a list of the mechanisms and their specific causes.
1. Mechanisms Leading to Decreased Cardiac Output (CO)
Cardiac Output is determined by Heart Rate (HR) x Stroke Volume (SV). Problems can arise with heart rate, contractility, or the volume of blood returning to the heart (preload).
A. Decreased Heart Rate (Bradycardia)
Mechanism: Inadequate heart rate reduces the number of times the heart pumps per minute.
Causes:
Vasovagal Syncope: A common cause of fainting due to a sudden drop in heart rate and BP.
Sick Sinus Syndrome: Malfunction of the heart's natural pacemaker.
Heart Block: Impaired electrical conduction in the heart.
Drugs: Beta-blockers, calcium channel blockers, digoxin.
Hypothyroidism: Severely low thyroid levels can slow the heart rate.
B. Decreased Stroke Volume
i. Reduced Preload (Inadequate Venous Return)
Mechanism: Not enough blood returning to the heart to pump out. This is the most common mechanism of hypotension.
Causes:
Hypovolemia (Low Blood Volume):
Hemorrhage (trauma, GI bleed)
Severe Dehydration (vomiting, diarrhea, burns, inadequate fluid intake)
Excessive diuresis (diuretics)
Venous Pooling:
Orthostatic (Postural) Hypotension: A failure of compensatory mechanisms upon standing.
Pregnancy: Pressure from the enlarged uterus on the inferior vena cava.
Varicose Veins
Drugs: Nitrates, opioids, some antidepressants.
Decreased Cardiac Filling:
Cardiac Tamponade (fluid around the heart)
Tension Pneumothorax (air in the chest cavity)
Superior Vena Cava (SVC) Syndrome
ii. Decreased Myocardial Contractility (Pump Failure)
Mechanism: The heart muscle is too weak to pump effectively.
Causes:
Myocardial Infarction (Heart Attack)
Cardiomyopathy
Heart Failure
Myocarditis (inflammation of the heart muscle)
Drugs: Beta-blockers, certain antiarrhythmics.
Severe Metabolic Derangements: Acidosis, hypoxemia.
iii. Increased Afterload (Obstruction to Outflow)
Mechanism: A physical obstruction makes it extremely difficult for the heart to eject blood.
Causes:
Massive Pulmonary Embolism
Aortic Stenosis (severe narrowing of the aortic valve)
Hypertrophic Obstructive Cardiomyopathy (HOCM)
2. Mechanisms Leading to Decreased Systemic Vascular Resistance (SVR)
This is a failure of the blood vessels to maintain adequate tone, causing widespread vasodilation.
A. Neurogenic Causes
Mechanism: A loss of sympathetic vasoconstrictor tone.
Causes:
Spinal Cord Injury (especially above T6) → Neurogenic Shock
General/Spinal Anesthesia
Severe Brain Trauma affecting the medullary cardiovascular centers
Autonomic Neuropathy (e.g., from diabetes, Parkinson's disease)
B. Septic Shock
Mechanism: The systemic inflammatory response to infection releases vasodilatory substances (like nitric oxide), causing profound vasodilation and capillary leak.
C. Anaphylactic Shock
Mechanism: A severe allergic reaction releases histamine and other mediators, causing massive vasodilation and increased capillary permeability.
D. Metabolic/Hormonal Causes
Mechanism: Underlying endocrine disorders or metabolic issues.
Causes:
Addison's Disease (Adrenal Insufficiency): Lack of aldosterone and cortisol leads to volume depletion and impaired vasoconstriction.
Severe Hypothyroidism: Can lead to decreased CO and blunted compensatory responses.
Severe Liver Failure: Leads to vasodilation and pooling of blood in the splanchnic circulation.
E. Pharmacologic Causes
Mechanism: Direct vasodilation from medications.
Causes:
Vasodilators (e.g., hydralazine, nitrates)
Alpha-blockers
Many antidepressants and antipsychotics
Alcohol
Summary Table of Major Types of Shock
The most severe form of hypotension is shock, which is categorized by the primary mechanism:
Type of Shock | Primary Mechanism | Common Causes |
|---|---|---|
Hypovolemic | ↓ Preload | Hemorrhage, dehydration |
Cardiogenic | ↓ Contractility | Heart attack, heart failure |
Obstructive | ↓ Preload or ↑ Afterload | Pulmonary embolism, cardiac tamponade |
Distributive | ↓ SVR (Vasodilation) | Sepsis, anaphylaxis, spinal cord injury |
In summary, hypotension arises from a disruption in the finely tuned systems that control heart function, blood volume, and vascular tone. Identifying the underlying mechanism is the first step toward effective treatment.
Describe the causes of hypertension and its relationship with the Renin-Angiotensin Aldosterone System.
Hypertension (high blood pressure) is a complex condition with multiple causes, and the Renin-Angiotensin-Aldosterone System (RAAS) is a central player in both its development and its consequences.
Part 1: Causes of Hypertension
Hypertension is classified into two main categories:
1. Primary (Essential) Hypertension
This accounts for 90-95% of all cases. There is no single identifiable cause; instead, it results from a complex interplay of genetic, environmental, and physiological factors.
Genetic Predisposition: Family history is a strong risk factor.
Lifestyle Factors:
Obesity: Increases blood volume, cardiac output, and often insulin resistance.
High Sodium Intake: Promotes water retention, increasing blood volume.
Excessive Alcohol Consumption
Physical Inactivity
Stress: Chronic stress can lead to sustained sympathetic nervous system activation.
Physiological Dysregulation:
Sympathetic Nervous System Overactivity: Increases heart rate, contractility, and vasoconstriction.
Renal Dysfunction: A reduced ability of the kidneys to excrete sodium, leading to volume expansion (the "sodium-volume hypothesis").
Vascular Inflammation and Endothelial Dysfunction: The endothelium (inner lining of blood vessels) loses its ability to produce vasodilators like nitric oxide, leading to increased vasoconstriction.
Hormonal Imbalances: Including abnormalities in the RAAS.
2. Secondary Hypertension
This accounts for 5-10% of cases and is caused by an identifiable underlying disease or agent.
Renal Disease:
Renal Artery Stenosis: Narrowing of the kidney arteries, which directly and powerfully activates the RAAS.
Chronic Kidney Disease: Impaired sodium and water excretion.
Endocrine Disorders:
Primary Aldosteronism: A tumor in the adrenal gland that overproduces aldosterone (e.g., Conn's syndrome), leading to sodium retention and potassium loss.
Pheochromocytoma: A tumor that secretes excessive catecholamines (epinephrine/norepinephrine), causing episodic or sustained hypertension.
Cushing's Syndrome: Excess cortisol, which has mineralocorticoid (aldosterone-like) effects.
Drug-Induced:
Nonsteroidal Anti-inflammatory Drugs (NSAIDs), decongestants, oral contraceptives, corticosteroids.
Other Causes:
Coarctation of the Aorta: A congenital narrowing of the aorta.
Sleep Apnea: Intermittent hypoxia increases sympathetic nervous system activity.
Part 2: Relationship with the Renin-Angiotensin-Aldosterone System (RAAS)
The RAAS is a critical hormonal system for regulating blood pressure, fluid, and electrolyte balance. When dysregulated, it becomes a major driver of hypertension.
The Normal RAAS Pathway:
Renin Release: The kidneys release renin in response to low blood pressure, low sodium, or sympathetic stimulation.
Angiotensinogen to Angiotensin I: Renin converts angiotensinogen (from the liver) to Angiotensin I.
Angiotensin I to Angiotensin II: Angiotensin-Converting Enzyme (ACE) in the lungs converts Angiotensin I to Angiotensin II, the primary effector of the system.
Aldosterone Release: Angiotensin II stimulates the adrenal glands to release Aldosterone.
How RAAS Activation Causes Hypertension:
The following chart illustrates how the overactive RAAS pathway leads to elevated blood pressure through its effects on blood vessels, the kidneys, and the nervous system:
In summary, an overactive RAAS raises blood pressure through multiple, synergistic mechanisms: intense vasoconstriction, expansion of blood volume, and potentiation of the sympathetic nervous system.
Specific Examples of the RAAS-Hypertension Relationship
In Primary Hypertension: Many patients have a normal or even low renin level. However, their vascular smooth muscle may be hyper-responsive to normal levels of Angiotensin II. A subset of patients (e.g., younger hypertensives) do have high-renin hypertension, where excessive SNS drive leads to elevated renin.
In Renal Artery Stenosis: The classic example of RAAS-driven hypertension. The narrowed artery reduces perfusion to the kidney, which is misinterpreted as low systemic blood pressure. This triggers a massive, inappropriate release of renin, leading to severe secondary hypertension.
In Primary Aldosteronism: Here, the problem is downstream of renin. Aldosterone is produced autonomously, independent of the RAAS feedback loop. This causes sodium retention, volume expansion, and hypertension, while suppressing renin release (low-renin hypertension).
The central role of RAAS in hypertension is why ACE inhibitors and Angiotensin Receptor Blockers (ARBs) are first-line and highly effective treatments. They directly target this pathway to lower blood pressure and reduce its damaging effects on the heart and kidneys.
List the drugs used in the treatment of hypertension and their targets.
The drugs used to treat hypertension target specific physiological mechanisms that control blood pressure, primarily focusing on reducing cardiac output (CO) and/or systemic vascular resistance (SVR).
Here is a list of the major classes of antihypertensive drugs, organized by their primary site or mechanism of action.
1. Drugs that Reduce Systemic Vascular Resistance (SVR)A. Act on the Renin-Angiotensin-Aldosterone System (RAAS)
1. ACE Inhibitors (Angiotensin-Converting Enzyme Inhibitors)
Examples: Lisinopril, Enalapril, Ramipril
Target: Angiotensin-Converting Enzyme (ACE).
Mechanism: Block the conversion of Angiotensin I to Angiotensin II (a potent vasoconstrictor) and decrease the breakdown of bradykinin (a vasodilator). The result is vasodilation and reduced aldosterone release.
2. ARBs (Angiotensin II Receptor Blockers)
Examples: Losartan, Valsartan, Irbesartan
Target: Angiotensin II Type 1 (AT1) receptors.
Mechanism: Block the action of Angiotensin II at its primary receptor, preventing vasoconstriction and aldosterone release. This leads to vasodilation.
3. Direct Renin Inhibitors
Example: Aliskiren
Target: Renin enzyme.
Mechanism: Directly inhibits renin, the first and rate-limiting step in the RAAS pathway, reducing the production of all downstream components.
B. Act Directly on Vascular Smooth Muscle
4. Calcium Channel Blockers (CCBs)
Examples (Dihydropyridines): Amlodipine, Nifedipine
Target: L-type calcium channels in vascular smooth muscle.
Mechanism: Block calcium influx into smooth muscle cells, preventing contraction and causing vasodilation. (Note: Non-dihydropyridine CCBs like Verapamil and Diltiazem act more on the heart).
C. Act on the Sympathetic Nervous System (Vascular Effects)
5. Alpha-1 Blockers
Examples: Doxazosin, Prazosin, Terazosin
Target: α₁-adrenergic receptors on vascular smooth muscle.
Mechanism: Block the action of norepinephrine, preventing sympathetic-mediated vasoconstriction and leading to vasodilation.
2. Drugs that Reduce Cardiac Output (CO)
Cardiac Output is reduced by decreasing Heart Rate, Contractility, or Blood Volume.
A. Reduce Heart Rate and Contractility
6. Beta-Blockers (β-Adrenoceptor Antagonists)
Examples: Metoprolol, Atenolol, Bisoprolol
Target: β₁-adrenergic receptors in the heart.
Mechanism: Reduce heart rate (negative chronotropy) and contractility (negative inotropy), thereby decreasing cardiac output. They also reduce renin release.
7. Non-Dihydropyridine Calcium Channel Blockers
Examples: Verapamil, Diltiazem
Target: L-type calcium channels in the heart and vasculature.
Mechanism: Act on the SA and AV nodes to reduce heart rate and conduction velocity. They also cause some vasodilation.
B. Reduce Blood Volume (Diuretics)
Diuretics increase sodium and water excretion by the kidneys, reducing plasma volume and preload, which decreases cardiac output.
8. Thiazide Diuretics
Examples: Hydrochlorothiazide (HCTZ), Chlorthalidone, Indapamide
Target: Distal Convoluted Tubule in the kidney.
Mechanism: Block the Na⁺/Cl⁻ cotransporter, promoting Na⁺ and water excretion. First-line for many patients.
9. Loop Diuretics
Examples: Furosemide, Bumetanide, Torsemide
Target: Thick Ascending Loop of Henle in the kidney.
Mechanism: Block the Na⁺-K⁺-2Cl⁻ cotransporter, causing a powerful diuresis. Used in more severe hypertension or with heart failure.
10. Potassium-Sparing Diuretics
Examples: Spironolactone, Eplerenone, Amiloride
Target: Collecting Duct in the kidney.
Mechanism: Spironolactone/eplerenone block aldosterone receptors, while amiloride blocks sodium channels. They cause mild diuresis while conserving potassium.
3. Drugs that Act Centrally on the Sympathetic Nervous System
11. Central Alpha-2 Agonists
Examples: Clonidine, Methyldopa
Target: α₂-adrenergic receptors in the brainstem.
Mechanism: Reduce sympathetic outflow from the central nervous system, leading to decreased heart rate and vasodilation.
4. Direct Vasodilators (Older Agents)
12. Arteriolar Dilators
Examples: Hydralazine, Minoxidil
Target: Vascular smooth muscle (via potassium channel opening, etc.).
Mechanism: Cause direct relaxation of arteriolar smooth muscle, leading to a marked reduction in SVR. Often used in resistant hypertension.
Summary Table of Drug Classes and Primary Targets
Drug Class | Primary Target | Main Physiological Effect |
|---|---|---|
ACE Inhibitors | Angiotensin-Converting Enzyme | ↓ Angiotensin II → Vasodilation |
ARBs | Angiotensin II Receptor (AT1) | ↓ Vasoconstriction → Vasodilation |
CCBs (Dihyd.) | Vascular Ca²⁺ Channels | ↓ Vasoconstriction → Vasodilation |
Thiazide Diuretics | Kidney (Distal Tubule) | ↓ Blood Volume → ↓ Cardiac Output |
Beta-Blockers | Cardiac β₁-receptors | ↓ Heart Rate & Contractility → ↓ CO |
Alpha-1 Blockers | Vascular α₁-receptors | ↓ Vasoconstriction → Vasodilation |
Loop Diuretics | Kidney (Loop of Henle) | ↓↓ Blood Volume → ↓ Cardiac Output |
Aldosterone Antagonists | Aldosterone Receptor | ↓ Blood Volume, ↓ Fibrosis |
Central Agonists | Brain α₂-receptors | ↓ Sympathetic Outflow |
Direct Vasodilators | Vascular Smooth Muscle | Direct Vasodilation |
In modern practice, ACE inhibitors, ARBs, CCBs, and thiazide diuretics are most commonly used as first-line agents, often in combination, as recommended by guidelines like those from the American Heart Association. The choice of drug is tailored to the individual patient's age, race, and co-existing conditions (e.g., diabetes, heart failure).
Describe the mechanism of neurohumoral activation in heart failure.
The mechanism of neurohumoral activation in heart failure is a classic example of a maladaptive feedback loop. Initially activated as a compensatory mechanism to support cardiac output and perfusion, it ultimately becomes destructive and drives the progression of the disease.
Core Concept: The Vicious Cycle of Heart Failure
In heart failure, the heart's ability to pump blood is impaired. The body misinterprets this as a sign of low blood volume and pressure, triggering powerful neurohumoral systems that are designed to correct this. However, in the context of a failing heart, these responses increase the heart's workload and cause further damage, creating a self-perpetuating "vicious cycle."
The following chart illustrates this self-reinforcing, maladaptive cycle:
Detailed Breakdown of the Key Systems1. Sympathetic Nervous System (SNS) Activation
Stimulus: Reduced cardiac output leads to unloading of arterial baroreceptors (in carotid sinus/aorta) and cardiopulmonary receptors. This reduces their inhibitory signals to the brainstem, leading to a surge in sympathetic outflow.
Acute Compensatory Effects (Initially "Helpful"):
Heart: ↑ Heart rate (chronotropy) and ↑ contractility (inotropy) to boost cardiac output.
Vessels: Vasoconstriction in non-essential beds (skin, gut, kidneys) to maintain blood pressure and shunt blood to the heart and brain.
Kidneys: Stimulates renin release (see RAAS below).
Chronic Detrimental Effects (The "Harm"):
Tachycardia and Increased Contractility: Increase myocardial oxygen demand, potentially leading to ischemia.
β₁-Adrenergic Receptor Downregulation: Chronic high catecholamine levels cause a decrease in the number and sensitivity of β₁-receptors on cardiac myocytes, making the heart less responsive to sympathetic stimulation and further compromising contractility.
Direct Cardiotoxicity: Norepinephrine and epinephrine can promote myocyte apoptosis (programmed cell death), hypertrophy, and fibrosis.
Arrhythmogenesis: Creates an electrically unstable heart, increasing the risk of sudden cardiac death.
2. Renin-Angiotensin-Aldosterone System (RAAS) Activation
Stimulus:
Renal Hypoperfusion: Decreased blood flow to the kidneys due to low CO and SNS-induced renal vasoconstriction.
Sympathetic Stimulation: Direct β₁-adrenergic stimulation of juxtaglomerular cells in the kidney.
Decreased Sodium Delivery: To the macula densa in the distal tubule.
Detrimental Effects:
Angiotensin II (AII):
Potent Vasoconstriction: Worsens afterload, making it even harder for the failing heart to eject blood.
Stimulates Aldosterone Release: Promotes sodium and water retention by the kidneys, leading to volume overload and edema.
Stimulates SNS & Vasopressin: Further potentiates the harmful neurohumoral cascade.
Promotes Remodeling: Directly causes cardiac and vascular hypertrophy and fibrosis.
Aldosterone: In addition to salt/water retention, it also promotes myocardial fibrosis, arrhythmias, and endothelial dysfunction.
3. Other Hormonal Systems
Vasopressin (Antidiuretic Hormone - ADH) Activation:
Stimulus: Baroreceptor unloading and Angiotensin II.
Effect: Acts on the kidneys (V2 receptors) to cause water retention, contributing to hyponatremia and volume overload. Also causes vasoconstriction (V1 receptors), increasing afterload.
Suppression of Natriuretic Peptides:
The heart secretes Atrial Natriuretic Peptide (ANP) and Brain Natriuretic Peptide (BNP) in response to stretch. They counteract the harmful systems by promoting:
Vasodilation (↓ SVR)
Sodium and water excretion (↓ volume)
Inhibition of Renin and Aldosterone
In chronic HF, although BNP levels are high, their effects are overwhelmed by the powerful SNS and RAAS, a state known as "NP resistance."
The Consequences: Cardiac Remodeling
The sustained activation of these neurohumoral systems leads to structural and functional changes in the heart, known as remodeling.
Myocyte Hypertrophy: Individual muscle cells enlarge.
Myocyte Apoptosis: Programmed cell death of muscle cells.
Interstitial Fibrosis: Stiffening of the heart muscle due to scar tissue.
Changes in Chamber Geometry: The ventricles become dilated and more spherical, which is mechanically less efficient.
This remodeling further impairs the heart's contractile function and pumping efficiency, perpetuating the cycle of neurohumoral activation and disease progression.
Clinical Implication
Understanding this mechanism is the basis for modern heart failure pharmacotherapy. Drugs like Beta-blockers, ACE inhibitors/ARBs, and Mineralocorticoid Receptor Antagonists are cornerstone therapies because they directly antagonize these maladaptive neurohumoral pathways, slow remodeling, and improve survival.
Describe the cardiovascular responses to exercise and acute stress (fight-or-flight response and vasovagal syncope).
The cardiovascular responses to exercise and acute stress are finely tuned adaptations to meet the body's changing demands. While the "fight-or-flight" response and exercise share similarities, they have distinct characteristics. Vasovagal syncope is a unique, paradoxical response to certain stressors.
1. Cardiovascular Response to Exercise
The primary goal during exercise is to dramatically increase cardiac output (CO) to deliver more oxygen and nutrients to working skeletal muscles and to remove metabolic waste. This is achieved through a highly coordinated series of events.
Central Command & Anticipatory Response
Even before muscle contraction begins, the cerebral cortex (central command) activates the autonomic nervous system, leading to an initial rise in heart rate and blood pressure.
Integrated Cardiovascular Adjustments
Heart:
Heart Rate (HR): Increases linearly with exercise intensity. This is due to withdrawal of parasympathetic (vagal) tone at low intensities and increased sympathetic stimulation at higher intensities.
Contractility: Sympathetic stimulation (via β₁-adrenoceptors) significantly increases the force of ventricular contraction.
Cardiac Output (CO = HR x Stroke Volume): Can increase 4-6 fold in a healthy individual. Both HR and Stroke Volume contribute, with SV plateauing at moderate intensities.
Circulation:
Massive Vasodilation in Skeletal Muscle: Local metabolic factors (e.g., hypoxia, adenosine, K⁺, H⁺, CO₂) override sympathetic vasoconstrictor tone, leading to a profound drop in vascular resistance in active muscle beds.
Vasoconstriction in Non-Essential Beds: Sympathetic outflow causes strong vasoconstriction in the splanchnic (gut), renal, and cutaneous circulations. This redirects blood flow away from these organs and toward the working muscles.
Systolic Blood Pressure (SBP): Increases significantly due to the rise in cardiac output.
Diastolic Blood Pressure (DBP): Remains relatively stable or decreases slightly because the massive vasodilation in muscle beds lowers total systemic vascular resistance (SVR).
Mean Arterial Pressure (MAP): Increases moderately.
Summary of Exercise Response: ↑↑ Heart Rate, ↑↑ Contractility, ↑↑ Cardiac Output, ↑ Systolic BP, ↔/↓ Diastolic BP. It is a balanced response that prioritizes blood flow to muscles while maintaining adequate perfusion pressure.
2. Cardiovascular Response to Acute Stress (Fight-or-Flight)
The goal of the fight-or-flight response is to prepare the body for immediate, intense physical activity to survive a perceived threat. It is mediated by a massive sympathetic nervous system (SNS) discharge and the release of catecholamines (epinephrine/norepinephrine).
Integrated Cardiovascular Adjustments
Heart:
Heart Rate (HR): Marked tachycardia.
Contractility: Sharply increased.
Cardiac Output (CO): Increases dramatically.
Circulation:
Vasoconstriction: Widespread and potent α-adrenergic mediated vasoconstriction in the skin, gut, and kidneys. This elevates Systemic Vascular Resistance (SVR) and shunts blood to the heart, brain, and prepared skeletal muscles.
Vasodilation in Skeletal Muscle: β₂-adrenergic stimulation from circulating epinephrine causes vasodilation in skeletal muscle vascular beds, preparing them for action. The net effect on muscle blood flow is a balance between metabolic demands and this neurogenic control.
Blood Pressure:
Systolic BP: Increases sharply due to increased CO.
Diastolic BP: Often increases due to the dominant effect of widespread α-adrenergic vasoconstriction (↑ SVR).
Mean Arterial Pressure (MAP): Significantly elevated.
Summary of Fight-or-Flight Response: ↑↑ Heart Rate, ↑↑ Contractility, ↑↑ Cardiac Output, ↑↑ Systolic BP, ↑ Diastolic BP. It is a generalized state of high alert with a primary goal of raising blood pressure to ensure cerebral and myocardial perfusion.
3. Cardiovascular Response in Vasovagal Syncope
Vasovagal syncope (the common faint) is a paradoxical and maladaptive response to certain stressors (e.g., emotional distress, pain, standing too long). Instead of preparing for fight-or-flight, the body initiates a "shutdown" response.
The Sequence of Events (Bezold-Jarisch Reflex)
Trigger: Often, a period of venous pooling (e.g., standing) leads to reduced venous return.
Compensatory Effort: The heart attempts to compensate with vigorous contractions.
Paradoxical Response: The underfilled, vigorously contracting ventricle activates mechanoreceptors (C-fibers) in the inferoposterior wall of the heart. These receptors mistakenly signal a state of high pressure to the brainstem.
Autonomic Storm:
Massive Increase in Parasympathetic (Vagal) Outflow: This leads to a profound bradycardia (slow heart rate) or even asystole (pause).
Massive Withdrawal of Sympathetic Outflow: This leads to a sudden and dramatic vasodilation and a drop in systemic vascular resistance (SVR), causing hypotension.
Summary of Vasovagal Syncope: ↓↓ Heart Rate (Bradycardia), ↓↓ Systemic Vascular Resistance (Vasodilation). The combination of a low heart rate and low peripheral resistance causes a catastrophic drop in blood pressure and cerebral perfusion, leading to a temporary loss of consciousness (fainting).
Comparison Table
Parameter | Dynamic Exercise | Fight-or-Flight Response | Vasovagal Syncope |
|---|---|---|---|
Heart Rate | ↑↑ | ↑↑ | ↓↓ (Bradycardia) |
Contractility | ↑↑ | ↑↑ | Variable |
Cardiac Output | ↑↑ | ↑↑ | ↓↓ |
Systemic Vascular Resistance | ↓ (in muscle), ↑ (elsewhere); Net: ↓ | ↑↑ (widespread vasoconstriction) | ↓↓ (Massive Vasodilation) |
Systolic BP | ↑↑ | ↑↑ | ↓↓ |
Diastolic BP | ↔ or ↓ | ↑ | ↓↓ |
Primary Control | Central Command & Local Metabolites | Sympathetic & Catecholamines | Parasympathetic & Sympathetic Withdrawal |
Goal / Outcome | Match O₂ Supply to Demand | Prepare for Immediate Action | Transient Loss of Consciousness (Faint) |
Define autoregulation of blood flow, reactive hyperemia and active (functional) hyperemia.
These are three fundamental concepts in local blood flow control that ensure tissues receive an adequate blood supply matched to their metabolic needs.
1. Autoregulation of Blood Flow
Definition: The intrinsic ability of an organ or tissue to maintain a relatively constant blood flow despite changes in perfusion pressure (the arterial pressure driving blood into the tissue).
The Problem it Solves: Arterial blood pressure is not constant; it rises and falls with daily activities. Without autoregulation, blood flow to an organ would passively increase when BP is high and decrease when BP is low, leading to potential damage or underperfusion.
The Goal: To ensure stable nutrient delivery and washout of waste products, independent of short-term fluctuations in systemic blood pressure.
Mechanisms (Two primary theories):
Myogenic Mechanism: Vascular smooth muscle in the arteriole wall contracts in response to an increase in pressure (stretch) and relaxes in response to a decrease in pressure. This is a direct response of the vessel to stretch.
Metabolic Mechanism: When pressure and flow decrease, oxygen delivery drops and metabolic waste products (CO₂, H⁺, adenosine, K⁺) accumulate. These metabolites cause vasodilation of the arterioles, restoring flow. When pressure is high, the washout of these metabolites allows the vessels to constrict.
Example: The kidneys and brain are excellent at autoregulation. If your blood pressure rises from 90 mmHg to 130 mmHg, the blood flow through your brain remains nearly constant because the cerebral arterioles constrict in response to the increased pressure.
2. Reactive Hyperemia
Definition: The dramatic increase in blood flow above resting levels that occurs following a temporary interruption of the blood supply (ischemia).
The Cause: The occlusion of an artery, which stops blood flow and creates a local metabolic crisis.
The Mechanism:
During the period of occlusion, oxygen levels plummet, and vasodilator metabolites (e.g., adenosine, CO₂) accumulate because they cannot be washed away.
When the occlusion is released, the high concentration of these metabolites causes powerful vasodilation of the downstream arterioles.
The previously ischemic tissue is now perfused at a much higher rate to "repay the oxygen debt" and clear the accumulated wastes.
Example: When a blood pressure cuff is inflated on the upper arm to a pressure above systolic pressure, the forearm becomes pale and cold (ischemic). When the cuff is released, the forearm flushes a bright red and feels warm as a wave of blood rushes in (reactive hyperemia).
3. Active (Functional) Hyperemia
Definition: The increase in blood flow to a tissue that is directly proportional to the increase in its metabolic activity.
The Cause: An increase in tissue function and metabolism (e.g., muscle contraction, gland secretion, neuronal activity).
The Mechanism:
Increased metabolic activity leads to a rapid increase in the consumption of O₂ and nutrients and the production of CO₂, H⁺, and other metabolites.
The relative lack of oxygen and the buildup of these vasodilator metabolites cause relaxation of the precapillary sphincters and arterioles in the active tissue.
This vasodilation increases blood flow, delivering more oxygen and nutrients and washing out the waste products, precisely matching the new level of demand.
Example: During exercise, blood flow to skeletal muscles can increase more than 20-fold. The contracting muscles consume O₂ and produce CO₂ and lactic acid, which dilate the arterioles, allowing more blood to flow in. Similarly, after a meal, blood flow to the stomach and intestines increases to support digestion.
Summary Table
Feature | Autoregulation | Reactive Hyperemia | Active Hyperemia |
|---|---|---|---|
Primary Stimulus | Change in Perfusion Pressure | Temporary Occlusion / Ischemia | Increase in Metabolic Activity |
Main Goal | Maintain constant flow during pressure changes | Repay oxygen debt after ischemia | Match blood flow to metabolic demand |
Key Metabolites | Changes in O₂/waste levels secondary to pressure change | Accumulation of metabolites (e.g., adenosine) during ischemia | Increased production of metabolites (e.g., CO₂, K⁺, adenosine) during activity |
Typical Scenario | Blood pressure rising or falling | Releasing a tourniquet | Exercising a muscle |
Describe the blood flow regulation mechanisms in major vascular beds of the body such as: renal, cerebral, and coronary circulations.
The regulation of blood flow is uniquely tailored to the function of each organ. Here is a description of the mechanisms in the renal, cerebral, and coronary circulations, which are among the most critically regulated vascular beds in the body.
1. Renal Circulation
Primary Function: Filtration of plasma to form urine, regulation of blood volume, electrolyte composition, and long-term blood pressure.
Key Characteristics:
Very High Blood Flow: Receives ~20-25% of cardiac output at rest.
High Oxygen Extraction: Despite high flow, it has high oxygen consumption due to active reabsorption processes.
Two Capillary Beds in Series: Glomerular capillaries (high pressure for filtration) and peritubular capillaries (low pressure for reabsorption).
Regulatory Mechanisms:
1. Strong Autoregulation:
Goal: To maintain a constant Glomerular Filtration Rate (GFR) despite fluctuations in systemic blood pressure (between ~80-180 mmHg mean arterial pressure).
Mechanisms:
Myogenic Mechanism: An increase in perfusion pressure causes stretch-induced constriction of the afferent arteriole, preventing a rise in pressure within the glomerulus.
Tubuloglomerular Feedback (TGF): Specialized cells (macula densa) in the distal tubule sense NaCl concentration. High flow and NaCl delivery signal constriction of the afferent arteriole to reduce GFR back to normal.
2. Hormonal Regulation (Extrinsic):
Sympathetic Nervous System (SNS):
Mild-Moderate Activation: Causes vasoconstriction of both afferent and efferent arterioles (via α₁-adrenoceptors), reducing renal blood flow (RBF) and GFR to shunt blood to other organs during stress or exercise.
Strong Activation (e.g., hemorrhage): Can cause severe vasoconstriction, dramatically reducing RBF to preserve central blood pressure.
Renin-Angiotensin-Aldosterone System (RAAS):
Angiotensin II: A potent vasoconstrictor, with a greater effect on the efferent arteriole. This unique action helps to maintain GFR even when renal perfusion pressure is low, by increasing the pressure within the glomerulus.
3. Other Humoral Factors:
Atrial Natriuretic Peptide (ANP): Released in response to volume overload, it relaxes the afferent arteriole and constricts the efferent arteriole, increasing RBF and GFR to promote sodium and water excretion.
Summary: Renal flow is dominated by autoregulation to protect GFR, but can be overridden by powerful sympathetic and hormonal inputs (SNS, Angiotensin II) to serve the body's systemic needs.
2. Cerebral Circulation
Primary Function: Provide a constant supply of oxygen and glucose to neurons and remove metabolic wastes. The brain is intolerant of ischemia.
Key Characteristics:
High Flow and Metabolic Rate: Receives ~15% of cardiac output.
Tight Blood-Brain Barrier (BBB): Protects the neural environment.
Limited Space: Enclosed within the rigid skull, so large changes in flow or volume can increase intracranial pressure.
Regulatory Mechanisms:
1. Powerful Autoregulation:
Goal: Maintain constant blood flow despite changes in systemic BP (effective between ~60-140 mmHg mean arterial pressure).
Mechanisms: Both myogenic and metabolic mechanisms are integral. The cerebral vessels are exquisitely sensitive to changes in perfusion pressure.
2. Metabolic Regulation (Most Potent):
Primary Signal: Changes in arterial CO₂ (and thus pH). An increase in PaCO₂ (hypercapnia) causes profound vasodilation, while a decrease (hypocapnia) causes vasoconstriction.
Other Signals: Hypoxia (low O₂) is a powerful vasodilator. Changes in neuronal activity lead to the local release of K⁺, adenosine, and H⁺, which fine-tune local flow to match metabolic demand (this is known as neurovascular coupling or functional hyperemia).
3. Neurogenic Control:
Relatively weak compared to metabolic control. Cerebral vessels are innervated by sympathetic nerves, which can provide vasoconstriction to protect against extreme increases in BP, and parasympathetic nerves, which can cause vasodilation.
4. Flow-Metabolism Coupling: Blood flow is tightly coupled to neuronal synaptic activity. An active brain region immediately experiences increased local blood flow.
Summary: Cerebral flow is exquisitely regulated by metabolic factors (especially PaCO₂) and powerful autoregulation to ensure a constant environment for neurons. It is relatively isolated from systemic hormonal changes.
3. Coronary Circulation
Primary Function: Supply oxygen and nutrients to the cardiac muscle itself. The heart has a very high and continuous metabolic demand.
Key Characteristics:
Phasic Flow Pattern: Blood flow to the left ventricle is highest during diastole and lowest during systole. This is because the powerful contraction during systole compresses the intramural coronary vessels, impeding flow.
High Oxygen Extraction: The heart extracts 70-80% of the oxygen from arterial blood at rest (compared to ~25% in most organs). To meet increased demand, it must increase flow, not extraction.
Regulatory Mechanisms:
1. Metabolic Regulation (Dominant):
Goal: Match blood flow precisely to the heart's metabolic rate (which is determined by heart rate, contractility, and wall stress).
Mechanism: Any imbalance between oxygen supply and demand leads to the accumulation of vasodilator metabolites. The most important are adenosine (a potent vasodilator), as well as K⁺, H⁺, CO₂, and lactate. A relative lack of oxygen (hypoxia) itself is also a direct vasodilator.
2. Autoregulation:
Very effective within a perfusion pressure range of ~60-150 mmHg. This ensures constant flow during the normal variations in aortic pressure that occur with each heartbeat and with daily activity.
3. Neurohumoral Regulation:
Sympathetic Nervous System:
Direct Effect: Norepinephrine acts on α₁-adrenoceptors on coronary vessels, causing vasoconstriction.
Indirect (Metabolic) Override: Sympathetic stimulation also increases heart rate and contractility, which massively increases metabolic demand and the production of vasodilators. The powerful metabolic vasodilation overrides the direct sympathetic constriction, leading to a net increase in coronary flow during exercise or stress.
Parasympathetic Nervous System (Vagus): Minimal direct effect, causing mild vasodilation via acetylcholine.
4. Endothelial Factors:
The coronary endothelium releases vasodilators like Nitric Oxide (NO) in response to shear stress (increased flow) and agonists (e.g., acetylcholine). This is crucial for flow-mediated dilation.
Summary: Coronary flow is predominantly regulated by local metabolic demand, with adenosine playing a key role. The phasic nature of flow and the high baseline oxygen extraction make it uniquely vulnerable to obstructions (e.g., coronary artery disease).
Comparison Table
Vascular Bed | Primary Regulator | Key Unique Feature | Response to Increased Activity |
|---|---|---|---|
Renal | Autoregulation (for GFR) | Tubuloglomerular Feedback | Flow decreases due to SNS activation (to shunt blood to muscles) |
Cerebral | Metabolic (PaCO₂, Neurovascular Coupling) | Blood-Brain Barrier | Flow increases locally in active brain regions |
Coronary | Metabolic (Adenosine, O₂ demand) | Phasic Flow (Diastolic Dominance) | Flow increases proportionally to cardiac work |
sympathetic innervation of blood vessels
SNS fibers innervate all vessels except capillaries and precapillary sphincters and some metarterioles.
precapillary sphincters are rings of smooth muscles that wrap around capillary beds that act as gatekeepers. (contraction= constriction=the sphincter closes off capillaries from blood flow) (relaxation= dilation= the sphincter opens capillary to blood flow)
metaarterioles provide a direct, low-resistance pathway for blood to flow from the arteriole to the venule, bypassing the true capillary network.
Large veins and the heart are sympathetically innervated.
-sympathetic nervous system innervation of small arteries and arterioles allows sympathetic nerves to increase vascular resistance by causing vasoconstriction.

what are the factors precapillary sphincters are controlled by?
Control: They are primarily controlled by local factors, such as:
Oxygen levels (low O₂ causes dilation “opening”).
Carbon dioxide levels (high CO₂ causes dilation “opening”).
pH (low pH/acidity causes dilation “opening”).
Metabolic byproducts (e.g., lactic acid, potassium ions).
This local control allows for autoregulation of blood flow, ensuring that the most active tissues receive the most blood.
how does the PSNS control the heart rate?
PSNS is mainly important in control of heart rate via the vagus nerve
sympathetic vasoconstrictor system (where is it located)?
remember, the sympathetic vasoconstrictor system constricts blood vessels to shunt blood to other places.
therefore, the sympathetic vasoconstrictor system would be in the kidneys, gut, spleen, skin (areas that don’t need as much blood when undergoing the fight or flight mode).
Receptor | Primary Effect on Vessels | Key Locations | Physiological Goal |
|---|---|---|---|
Beta-2 (β₂) | Vasodilation (epinephrine) | Skeletal Muscle, Liver, Heart | Increase blood flow to active tissues. |
Alpha-1 (α₁) | Vasoconstriction (norepinephrine) | Skin, GI Tract, Kidneys, Splanchnic Circulation | Increase blood pressure by raising SVR; shunt blood away from non-essential organs. |

what is the formula for arterial pressure?
Arterial Pressure ~ CO× SVR
svr: systemic vascular resistance
arterial pressure
Arterial Pressure ~ CO× SVR
svr: systemic vascular resistance
Arterial Pressure is the force exerted by the blood against the walls of the arteries. It is the driving force that propels oxygenated blood from the heart to the tissues and organs throughout the body.
1. Constricting almost all arterioles → Increases SVR
vasoconstriction is the most direct way to increase pressure because it makes the vessels narrower. Widespread constriction of arterioles (the primary resistance vessels) dramatically increases Systemic Vascular Resistance (SVR), which leads to an increase in arterial pressure
Physiological Example: The sympathetic nervous system does this via alpha-adrenergic receptors to prevent a drop in blood pressure when standing up (postural hypotension).
2. Constricting large vessels (veins) → Increases Venous Return, EDV, and CO
Mechanism: This is a more subtle but critically important mechanism. The largest volume of blood is in the venous system. Constricting these large veins (venoconstriction) reduces their capacity→ "squeezing" blood back toward the heart.
Why it Works:
Increased venous return increases End-Diastolic Volume (EDV)—the amount of blood in the ventricle just before contraction.
According to the Frank-Starling law, a greater EDV stretches the heart muscle, leading to a more forceful contraction and a greater Stroke Volume (SV).
Since CO = HR × SV, this increased SV directly leads to an increased CO.
Physiological Example: This is a key function of sympathetic nervous system activation during exercise or stress, ensuring the heart has enough blood to pump.
3. Directly increasing heart rate and contractility → Increases CO
Mechanism: The sympathetic nervous system directly acts on the heart.
Increasing Heart Rate ( Chronotropy): Via beta-1 receptors in the SA node.
Increasing Contractility ( Inotropy): Via beta-1 receptors in the ventricular myocardium, making each contraction more powerful and ejecting a larger Stroke Volume (SV).
Why it Works: Both of these actions directly increase Cardiac Output (CO = HR × SV). According to the formula AP = CO × SVR, if SVR remains constant, an increase in CO leads to a direct increase in Arterial Pressure.

what is the difference between vasoconstriction in arterioles, veins, and sympathetic directly increasing heart rate through the b1 receptor?
what is the similiarity?
vasoconstriction in arterioles (because they pump blood away) leads to higher SVR → higher AP
vasoconstriction in veins (because veins bring blood back) leads to increased venous return, edv and cardiac output via frank starling
sympathetic directly increases heart rate to increase CO → high AP
similarity: increased arterial pressure.