Lecture #9 & 10 | Parsimony Cladistics
1/35
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
36 Terms
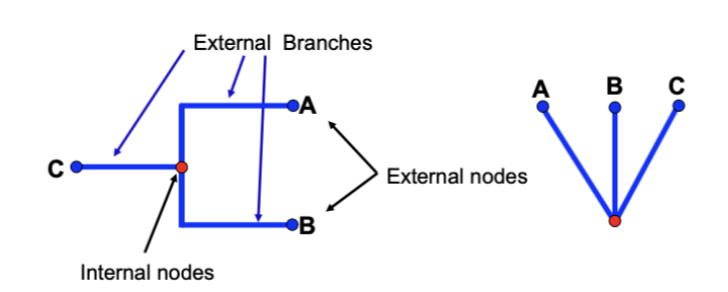
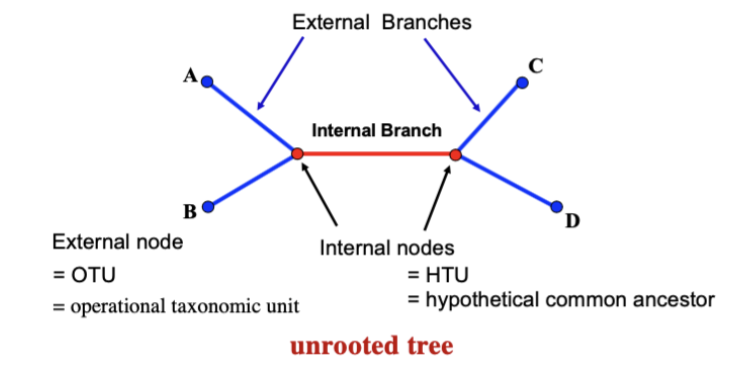
Internal nodes/External Branches and Nodes
Optimality Criterion (OC)
Uses optimality criteria to choose among set of possible trees
Advantage: OC methods require an explicit function for relating a tree to data
Disadvantage: Can be computationally expensive; the problem of finding the best tree from among all possible trees is difficult
Synapomorphy
Derived character state shared by two or more taxa and their common ancestor
Plesiomorphy
primitive or ancestral character state
Autapomorphy
Unique change not informative for relationships
Apomorphy
Any derived character state or feature within a lineage
Parisomony
The shortest hypothesis is the best, even though the alternative may be the correct hypothesis
Procedure or parsimony
selective suite of informative characters (from 1 to many)
code alternative character states
estimate based on the number of shared apomorphic states
beginning w/ the taxa that share the most derived character states, begin to add other taxa
How to select characters in parsimony
As many characters should be chosen
each character gives equal weight to the determination of overall similarity
How to manually create a parsimony
group taxa with most synapomorphies
add the next group with most synapomorphies
add remaining taxon and map all characters
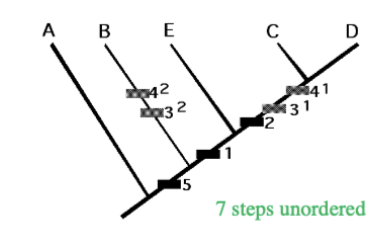
Fitch analysis
All character states unordered
Wagner analysis
All character ordered ( no difference if all binary)
General analysis
a mix of both ordered and unordered characters. Some characters may be better suited for ordering that others
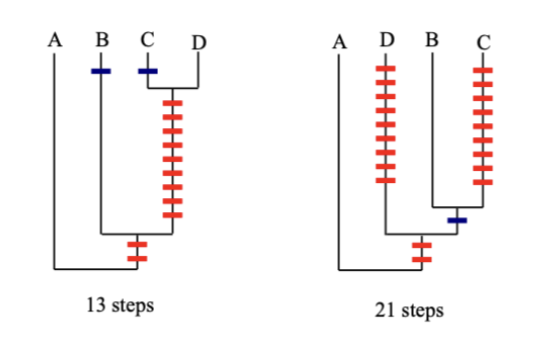
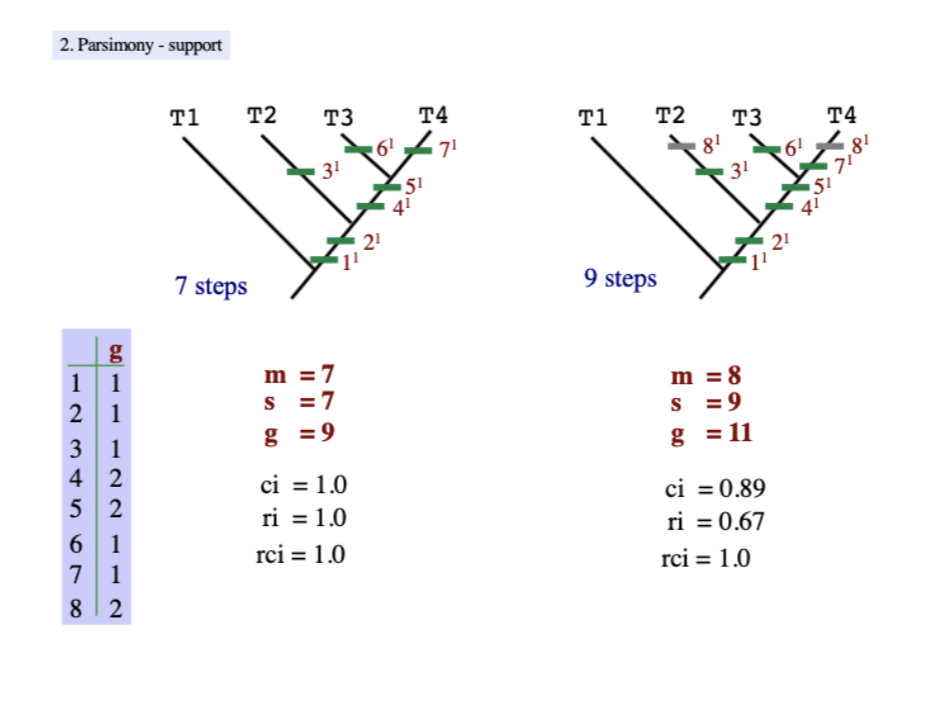
How do fitch and Wagner analysis compare in the same data
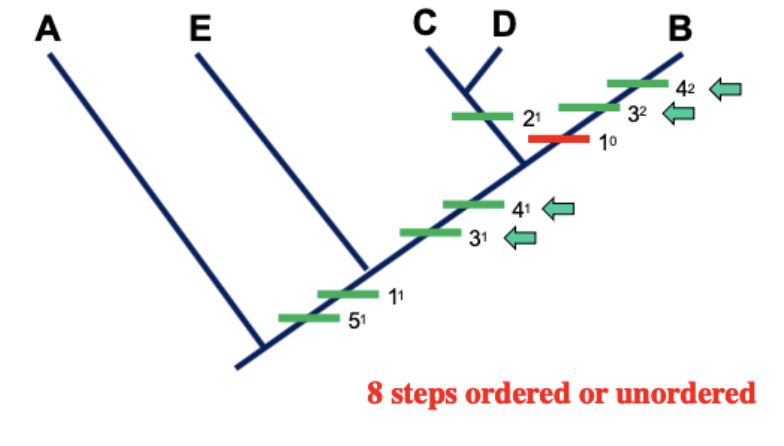
Notice that with Wagner, that same data takes more steps
Parsimony favors the tree that has the shortest length
Clarification of a “step” in cladistics
Each evolutionary novelty (apomorphy) is one step
Includes:
homoplasies (convergence and parallelism)
reversals
autapomorphies
Plesiomorphies and characters found in all taxa are not counted
Exhaustive search
A way to search for the most optimal tree by evaluating all trees
should be done for 10 or few taxa
Exact search
A way to search for the most optimal tree where all trees are not evaluated, but search is guaranteed to find the best tree
limited is less than 20 taxa
Uses a branch and bound algorithm that focuses on sets of trees
Heuristic searches
A way to search for the most optimal tree but is not guaranteed to find the best tree
searches are almost always heuristic with more than 20 taxa
Steps:
Build initial search tree
swap branches/taxa to search for a shorter tree
search and keep all trees of the same length
Nearest neighbor interchange (NNI)
A method of branch swapping that swaps the closest branches only
computationally fast
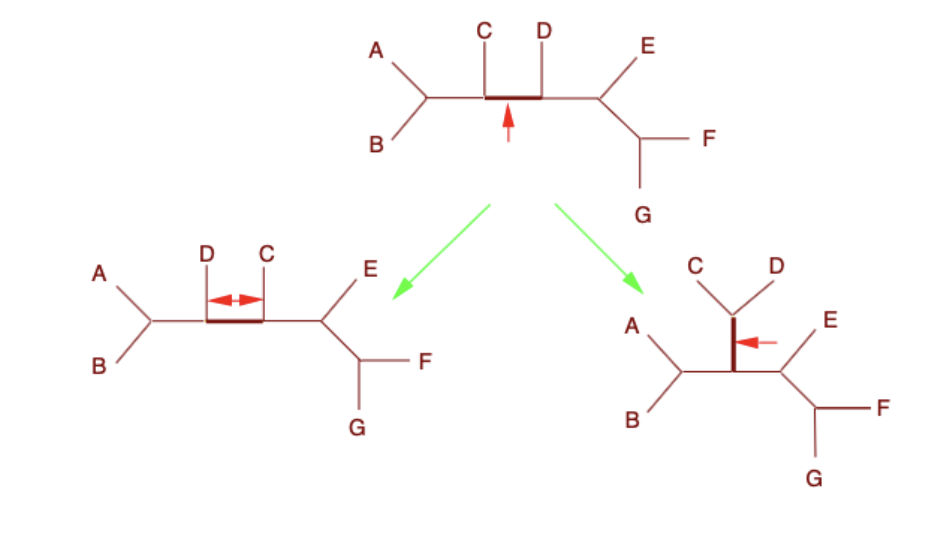
Subtree pruning and regrafting (SPR)
Intermediate changes grafting only at the point cut off
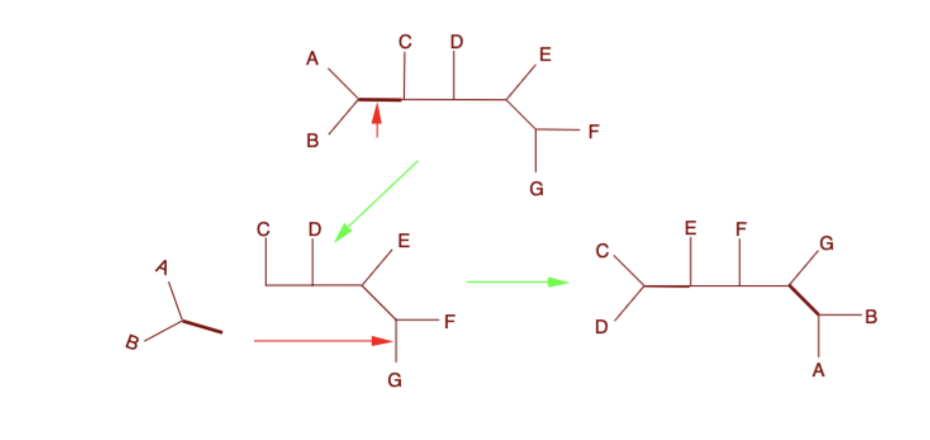
Tree bisection and reconnection (TBR)
Major changes involving cutting of groups and reattaching at any of the internal branch points
it is the most computationally expensive, but best for making rearrangements more likely to find the shortest tree
Most common method

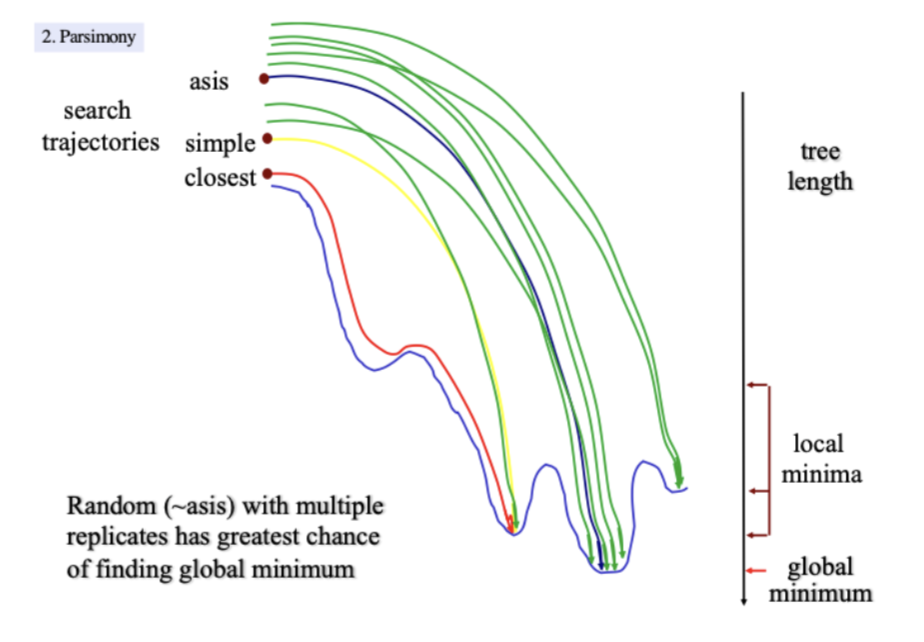
Heuristic strategies
build initial search tree (asis, closest, simple, random)
swap branches/taxa to search for a shorter tree
search and keep all trees of the same length
Stepwise addition in Heuristics
Asis: order in the data matrix
closest: starts with shortest 3-taxon tree adds taxa in order that produces the least increase in tree length
simple: the first taxon in the matrix is the reference
taxa are added to it in the order of their decreasing similarity to the reference
random: taxa are added in a a random sequence, many different sequences can be used
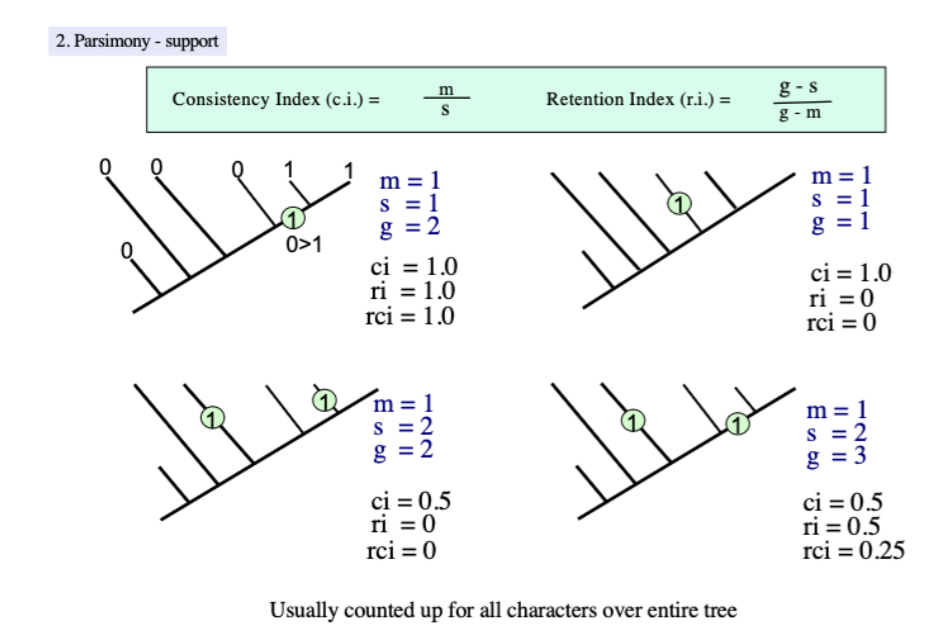
Consistency index (CI)
a measurement of tree strength that
equation: m/s
m=minimum steps
s=actual number of steps
Measures the amount of homoplasy on a particular tree
decreases with the number of evolutionary steps needed to explain the distribution of characters
1: A character has perfect consistency
Two state characters (0/1): CI value of 0.5
Retention index (RI)
measurement of the fit of a character or how much a character change contributes to a tree topology
(g-s)/(g-m)
m=minimum steps
s=actual number of steps
g= minimum number of any particular state on the tree
A character of perfect fit gets an RI of 1
An autapomorphy gets an RI of 0
Rescaled consistency index
ri x ci
A function to more adequately scale the value of the fit of a character (ri) to 0
trees that have a retention index of less than 0.7 may have multiple fundamentally different tree topologies that can be of the same length, so rigorous analyses are needed
Parsimony support examples
Problems of cladistics methods
choice of characters may influence results
chance of unresolved or conflicting nodes on the tree
homoplasy (convergence, parallelism, and reversals (loss of acquired traits))
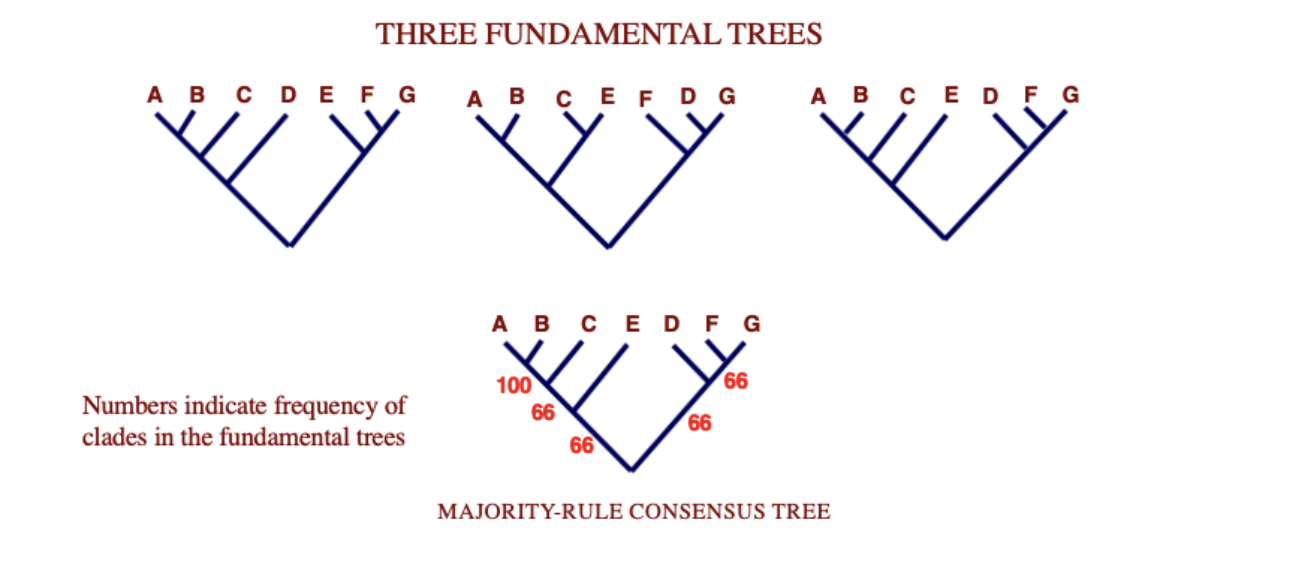
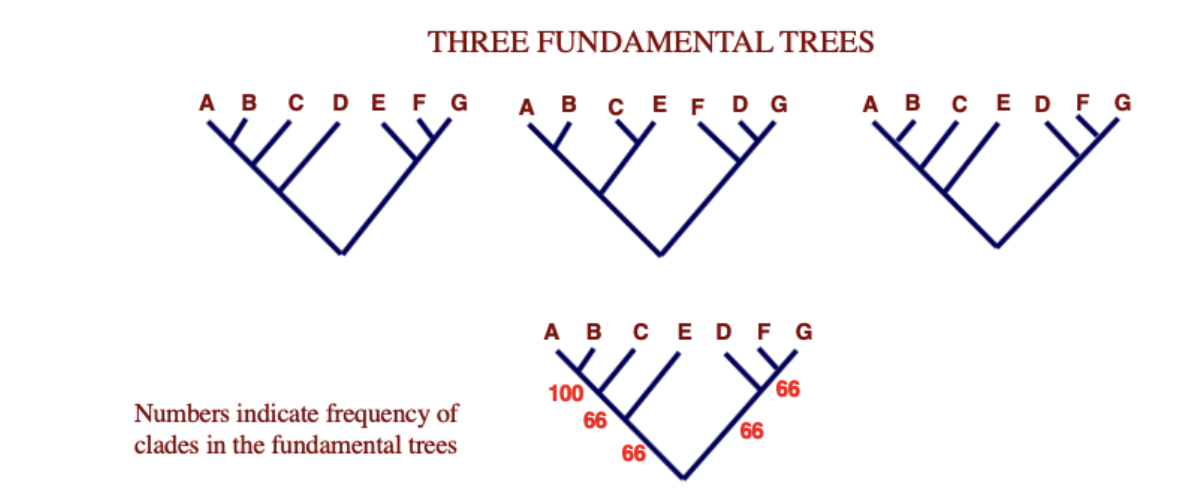
What happens if there are multiple trees that have the same “shortest” length
different trees can be combined together into a consensus tree
multiple due to
alternative equally parsimonious optimizations of homoplastic characters
missing data
or both
Most common relationships are summarizes with consensus trees

Strict consensus methods
only includes groups that appear in all of the input trees
most conservative model for consensus trees
Majority rule consensus tree
tree that includes clades that appear in more than half of a collection of trees
numbers indicate frequency of clades in the fundamental trees
Bootstrap
Commonly used measure of support for many of the OC methods of analysis
characters are resampled randomly from original data, with replacement, until new data set with original number of observations obtained
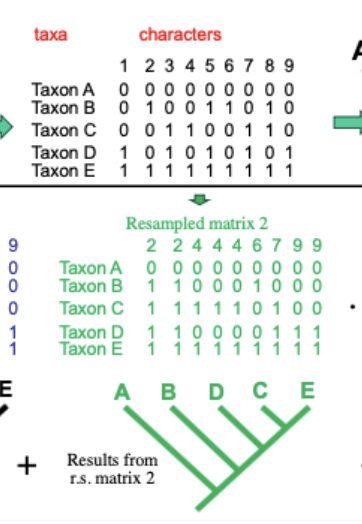
tree is computed for each replicated data set
agreement among the resulting trees is summarized with a majority-rule consensus tree
BP proportion: frequency of occurrence of a clade and is a measure of support for a group
Intersections (A ∩ B)
the same item found in each group

Note: work through problem in lecture 10
Union ( A ∪ B )
All items in each group


Note: work through problem in lecture 10
Advantages of parsimony
simple method - easily understood operation
does not depend on an explicit model of evolution
gives both trees and associated hypotheses of character
evolution
should give reliable results if the data is well structured and
homoplasy is either rare or widely (randomly) distributed on
the tree
Disadvantages
may give misleading results if homoplasy is common or concentrated in particular parts of the tree
underestimates branch lengths for molecular data
simple model of evolution, but is simple the best
parsimony often justified on philosophical groups
not compelling for molecular