(pt 2) exam #5 - heme II (cls 546)
1/85
Earn XP
Description and Tags
disorders of secondary hemostasis + acquired disorders w bleeding
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
86 Terms
goal of secondary hemostasis?
stable fibrin clot
2 categories of disorders in secondary hemostasis?
disorders of fibrin formation + disorders of fibrinolysis
symptoms associated with typical disorders of secondary hemostasis?
Delayed bleeding
Deep muscular bleeding
Joint bleeding
how does one "get" a disorder of fibrin formation?
Hereditary or acquired
Quantitative or qualitative
disorders of proteins of fibrin formation classifications
X-linked recessive
FVIII & FIX
Autosomal dominant (AD)
VWD ; dysfibrinogenemia
Autosomal recessive (AR)
All the rest
what are the expected coag screening results with disorders of secondary hemostasis? does this differentiate between qualitative and quantitative disorders?
Prolonged PT and/or APTT
No
what tests would you order after a prolonged PT and/or PTT?
Platelet count, fibrinogen (if not ordered already)
1:1 mix
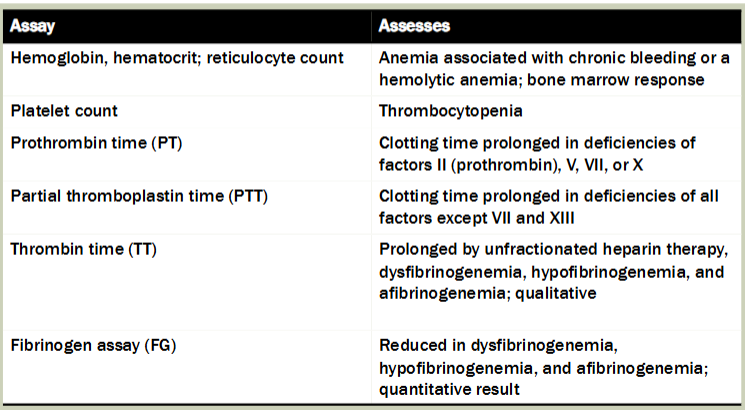
primary assays for generalized hemostatic disorders (chart)
indications for congenital bleeding disorders
Relatives with similar bleeding symptoms
Onset of bleeding in infancy or childhood
Excessive bleeding from umbilical cord or circumcision wound
Repeated hemorrhages in childhood, adulthood
Chronic petechiae, purpura, or ecchymoses
Bleeding into joints, central nervous system, soft tissues, peritoneum
von willebrand disease (VWD)
Inherited hemorrhagic disorder
Genetically and clinically heterogeneous
Caused by a deficiency or dysfunction of VWF
Most common hereditary bleeding disorder
125 in 1 mill have VWD w bleeding symptoms
Autosomal dominant in most patients
Autosomal recessive patients more severely affected
First described by Erik von Willebrand in 1926
(VWD) von willebrand factor
Synthesized in the endothelial cells & megakaryocytes
Mediates platelet adhesion
Complexes with and stabilizes FVIII
Carrier protein
Platelet adhesion with GpIb/IX/V
Binds to collagen and GPIB/IX
(VWD) synthesis and structure of VWF
Synthesized by Ecs
Multimeric chain of identical subunits
VWF molecules bind FVIII in 1:1 ratio
Defects in VWF may:
Interfere with ligand interaction
FVIII ; GPIB/IX
GPIIb/IIIa
Collagen ; Heparin
Cause bleeding by impairing either platelet adhesion or blood clotting
types of VWD
Types 1 & 3: quantitative deficiencies of VWF
Type 2: four subtypes ; qualitative deficiencies
type 1 & 3 VWD
BOTH ARE QUANTITATIVE DEFICIENCIES OF VWF
Type 1 (70-80% of cases)
Most common
Mild w partial deficiency of VWF
All multimers present but reduced
Type 3 (0.5-5 per million)
Absolute absence of VWF and VWF:Ag
Severe form of disease
type 2 VWD
Qualitative abnormalities of VWF
VWF activity is consistently reduced
May have normal levels of VWF protein but protein is dysfunctional
15-20% of cases
Four subtypes or variants
clinical manifestations of VWD
Usually mild bleeding symptoms which reflect a platelet problem
Epistaxis ; menorrhagia
Hemorrhage in mucosal and cutaneous tissues
Bleeding following tooth extraction
Gingival bleeding
Easy bruising
Homozygotes or double heterozygotes—more severe
Great phenotypic variability in symptoms and lab results
VWD vs hemophilia
NO hematoma and hemarthroses
Not prominent (unlike hemophilia A)
NO major hemorrhagic tendency
Absence of bleeding symptoms does not rule out this diagnosis
laboratory evaluation of VWD
Screening tests for fibrin formation
Do not directly evaluate VWF
Requires a battery of tests
Platelet count, PT, APTT, PFA-100
Specific tests
Quantitate VWF & FVIII activity
Determine various functional and structural aspects of VWF protein
Platelet aggregation studies
Remember we need both VWF:A and VWF:Ag for diagnosis!!
(additional testing for VWD) VWF:Ag
Cannot measure VWF:Ag by clot-based assays
Immunologic testing required to quantitate
Use monoclonal ABYs to VWF (ELISA), LIA
Patients ABO type affects level of VWF
Type O has 25-30% less than A and B
(additional testing for VWD) VWF:Rco
VWF ristocetin cofactor activity; aka VWF activity test
Functional assay ; quantitative
measured by the ability to cause agglutination of reagent platelets by the patient’s VWF
Agglutination is measured in an aggregometer
VWD laboratory diagnosis
RIPA—Ristocetin-induced platelet agglutination
Uses patients platelet rich plasma
Measures ability of ristocetin to induce agglutination of patient platelet rich plasma
Detects patient's VWF binding to patient's GPIb/IX
Abnormal in both BSS and VWD
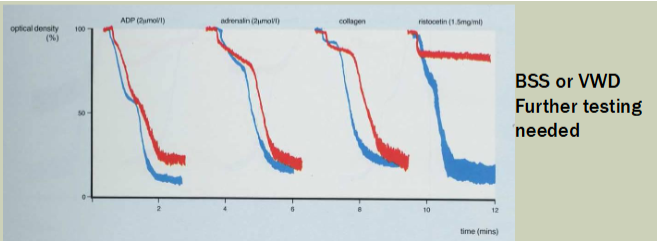
VWD vs FVII deficiency
APTT
Will be prolonged when FVIII activity is <30%
Often normal in VWD
FVIII activity
Measured using factor assay
Modified APTT
Uses human FVIII deficiency plasma as a substrate
Normal levels are 50-150%
VWD diagnosis (summary)
PFA 100 closure time
Uses collagen/ADP and COL/EPI to assess aggregation
Abnormal in VWD with both agents
Diagnosis of VWD
Decreased VWF:Ag, decreased VWF activity, decreased FVIII activity, and/or prolonged PFA closure time
Classic case: all four laboratory tests are abnormal

VWD lab diagnosis challenges
FVIII level is variably reduced in VWD
APTT
Typically prolonged only when FVIII <30%
Often normal in VWD
PT and TT are normal
VWF:Ag
Acute phase reactant
Blood type quandary (type has less Ag)
final step in VWD diagnosis / confirmation
Establish subtype using SDS agarose electrophoresis
Evaluate multimeric structure of VWF
Multimers are separated by size and visualized as bands
DNA analysis
Specific gene mutation on chromosome 12
Gene is large and complex with high degree of polymorphism--makes DNA analysis difficult
acquired von willebrand syndrome (AVWS)
Rare ; loss of VWF secondary to:
Neutralizing antibodies, protein degradation, adsorption to cell surfaces
Occurs in previously normal individuals
Present w new-onset bruising and bleeding
Reduced VWF activity, VWF:Ag and FVIII
Can see increased levels of VWF propeptide (VWFpp)
Associated with:
Underlying lymphoproliferative disorders (CLL/SLL, MM, MGUS etc) and autoimmune disorders
therapy for VWD
Goal: raise levels of VWF
Cryoprecipitate
Classic treatment; has molecular forms of VWF, FVIII, and fibrinogen
DDVAP (deamino-D-arginine-vasopressin or Desmopressin)
Preferred ; modified ADH
Induces endothelial cell release of VWF from Weibel-Palade bodies
Temporarily increases levels of VWF and FVIII
No effect in type 3 VWD patients
Concentrated "intermediate purity" FVIII products
Contain large amounts of intermediate size VWF molecules
VWF levels increase after administration
For nonresponders to DDVAP
hemophila (general)
X-linked recessive disorders
Females are carriers and pass abnormal X chromosome to sons who are affected
Hemophilia A—VIII deficiency
Antihemophilic factor
Hemophilia B—IX deficiency
Christmas factor
pathophysiology of hemophilia
Insufficient generation of thrombin by FIXa/VIIIa complex through intrinsic pathway of coagulation cascade
Results in:
Inadequate fibrin formation (no thrombin burst/propagation)
Excessive fibrinolysis due to lack of thrombin activation of TAFI
mutations in hemophilia
Occur throughout the genes for FVIII and IX
Males bearing single defective allele affected with hemophilia
Severity determined by site of mutation
Inherited from carrier mother or spontaneous mutation
Hemophilic males do not transmit genes to sons
All daughters are obligate carriers for hemophilia
(hemophilia) factor VIII gene mutations
Majority result in either quantitative or qualitative defects
Most are CRM or CRM^R (cross reacting material negative or reduced) ~95%
i.e. quantitative!!
Means there is a decrease or absent clotting activity by both functional and immunological testing
CRM+ = normal levels of a dysfunctional FVIII
Inversion mutation
Involves intron 22
Occurs in 50% of patients w severe disease
(hemophilia) factor IX gene mutations
Mutations in FIX gene or its regulatory components
Severity dependent on type and region of mutation
Mutations = mild -> mod -> severe hemophilia
1/3 are CRM+ (qualitative defect)
Have the normal quantities of a dysfunctional FIX
2/3 are CRM- or reduced (quantitative defect)
clinical aspects of hemophilia (levels of hemophilia)
Clinical severity corresponds with level of factor activity
>30% activity--no abnormal bleeding
Severe hemophilia
Factor coagulant activity < 1% of normal
Frequent spontaneous bleeding into joints and soft tissues
Prolonged bleeding with trauma or surgery
Moderate hemophilia
Factor coagulant activity 1–5% of normal
Occasional spontaneous bleeding
Mild hemophilia
Factor coagulant activity > 5% of normal
Rare spontaneous bleeding
severe hemophilia
Bleed from any anatomic site after negligible or unnoticed trauma
Most common symptoms
Hemarthrosis--chronic inflammation, deterioration
Intra-articular and intramuscular bleeds
Soft-tissue bleeds, bleeding associated with intramuscular injections and surgery, oral bleeding, hematuria, GI tract bleeding
Most common hemorrhagic cause of death: intercranial hemorrhage
how do hemophilia A and B present?
present the same ; diagnosis depends on:
Unusual bleeding symptoms early in life
Age of first bleeding varies with severity of disease
Family history
Physical exam
Laboratory evaluation
laboratory evaulation of hemophilia
PT, TT, and platelet function—normal
APTT prolonged
Mild deficiencies may not be detected (levels between 20 and 50 IU/dL or >30% activity
confirm w specific factor assays for FVIII and FIX
Molecular diagnosis--DNA probes
Use mixing studies to rule out inhibitor
(hemophilia) carrier detection
Challenging to detect
Cannot detect with plasma tests due to variability in expression
VIII carriers
VWF:Ag level to FVIII level—2:1 (more vWF than VIII)
Genetic testing is preferred method
Acute phase reactant ↑ w/exercise, stress, inflammation
1/3 arise from point mutations and cannot be predicted
Inversion of intron 22 (most common mutation)
IX carriers
Direct gene sequencing
therapy for hemophilia
Goal is replacement of clotting factor to achieve hemostasis
Hemophilia A
Heat or solvent-detergent treated cryoprecipitate preparation or FVIII
FVIII and FIX products
prepared using monoclonal antibodies
Recombinant DNA technology products are preferred
DDAVP—mild hemophilia A
Gene therapy—showing promising results in clinical trials
(hemophilia) inhibitors
Alloantibodies
Neutralize coagulant effects of replacement therapy
Seen in:
5-20% of hemophilia A patients
1-3% of hemophilia B patients
Most often produced by pts w large deletions
CRM- patients have higher risk of developing an inhibitor than CRM+ patients (quantitative deficiency vs qualitative)
what tests would be prolonged in Factor I (fibrinogen) deficiency?
PT/INR, APTT, TT
fibrinogen deficiency (general)
Quantitative defects--type I
Afibrinogenemia--23%
Hypofibrinogenemia--26%
Qualitative defect--type II
Dysfibrinogenemia--51%
Hypo-dysfibrinogenemia
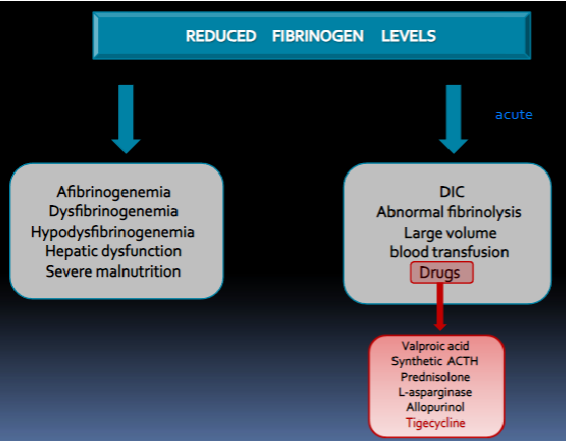
afibrinogenemia
No detectible fibrinogen by any method
Severe bleeding disorder but milder than severe hemophiliacs
PT, APTT, TT--abnormal
Correct by mixing studies
BT and plt agg studies are normal
Definitive diagnosis
Antigenic and functional assays--<1 mg/dL
Rule out heparin contamination, fibrinogen degradation (FDPs), inhibitor
Associated with recurrent pregnancy loss
Therapy = cryoprecipitate or fibrinogen concentrates
hypofibrinogenemia
50% of normal levels of fibrinogen
Milder bleeding course--often asymptomatic
Associated with recurrent pregnancy loss
acquired by:
Reduced/absent or abnormal fibrinogen synthesis
Fibrinogen loss exceeds fibrinogen production
Hyperfibrinolysis
dysfibrinogenemia
Normal fibrinogen levels but abnormal structure and function (qualitative issue)
Autosomal dominant--most often seen in heterozygous state
50% are asymptomatic
25% mild bleeding
25% thrombosis
Differentiate from acquired forms
Such as liver disease, pancreatitis, and so on
tests for dysfibrinogenemia
PT, APTT, and PFA—usually normal
TT, clot-based quantitative assay, reptilase time—abnormal (activity tests abnormal)
Antigenic fibrinogen assays normal
>600 mutations have been identified
Some involve:
The site of cleavage to fibrin (and/or release of FP A or FP B)
The site of polymerization of fibrin (no polymers)
The binding site for XIIIa cross linkage
Thrombin binding sites
prothrombin deficiency (FII)
Genetically heterogeneous
Hypothrombinemia--quantitative
Dysprothrombinemia--qualitative
Congenital prothrombin deficiency
Rare--1 in 2,000,000 (rarest of them all)
Homozygotes
<10% of normal--severe bleeding
Complete absence--incompatible with life
Heterozygotes for hypoprothrombinemia
Levels of ~50% of normal--asymptomatic
(prothombin deficiency) lab results
Prolonged PT, APTT
Normal TT and platelet function studies
(prothrombin deficiency) diagnosis + treatment
diagnosis
Specific factor assay
Immunologic tests for antigen
Exclude vitamin K deficiency
treatment: PCC--prothrombin complex concentrates
factor V deficiency
Disorders genetically heterogeneous (can be quantitative/qualitative)
Lab tests
PT and APTT prolonged ; TT normal
Diagnosis--specific FV assay
Therapy—fresh frozen plasma
Plt products can provide FV
factor VII deficiency
Only PT/INR is prolonged
Rare disorder; may be quantitative or qualitative
Homozygous--<10 U/dL
Heterozygous--40-60 U/dL
Definitive diagnosis
FVII assay--functional and quantitative
Therapy
Recombinant FVII (NovoSeven), prothrombin complex concentrates (PCC), FVII concentrates
Gene therapy
FVII deficiency symptoms
No symptoms (54%)
Bleeding after trauma (17%)
Mild, spontaneous bleeds, bruising, nose bleeds, abnormal menstrual periods (22%)
Major bleeds, joint bleeding, brain bleeds, stomach intestines, and umbilical cord (7%)
factor X deficiency
Rare disorder—quantitative or qualitative
Lab tests
PT, APTT, and russel viper venom test (directly activates X)--prolonged
Chromogenic assay / immunological assay
Platelet function test--normal
Definitive diagnosis—specific factor assay for FX
Therapy—fresh frozen plasma and prothrombin complex concentrate (PCC)
what are the assays that can measure factor X? (5)
PT-based
PTT-based
Chromogenic Factor X
Immunological Factor X
dRVVT
what is hemophilia C? what tests are prolonged in it?
Deficiency in factor XI
APTT
factor XI deficiency (hemophilia C)
occurs most often in Ashkenazi Jews
Autosomal recessive--affects males and females
Most are type I--quantitative
Lab tests—prolonged APTT, other tests are normal
Diagnosis--specific FXI assay
Treatment
FXI concentrates--available in Europe
FFP ; low dose rFVIIa
what test is used to detect Factor XIII deficiency?
5M urea solubility test
forms of factor XIII deficiency
highly heterogenous ; rare ; pts lack both plasma and platelet FXIII
Deficient of both subunits A and B (Type 1)
Deficiency of A subunit only (Type 2)
Deficiency of B subunit only (Type 3)
hallmarks of factor XIII deficiency
Umbilical stump bleeding and bleeding after circumcision
Intracranial hemorrhage with little or no trauma
Recurrent spontaneous abortion
Bleeding at time of surgery is not excessive
Delayed bleeding can occur
(factor XIII deficiency) lab results + diagnosis
Laboratory results
Normal PT, APTT, TT, and platelet function despite history of bleeding
Abnormal in vitro clot formation
Diagnosis
Solubility of fibrin clots in 5M urea
Needs <1% of XIII activity to demonstrate deficiency
Specific assays for FXIII available
you have a normal PT but a prolonged PTT with no history of bleeding, what factors would you suspect have a deficiency?
FXII, PK, HK
combined factor deficiencies
Vitamin K deficiency (II, VII, IX, X)
Factors are produced by no gamma carboxylation (non-functional)
PT/PTT prolonged, TT normal
Combined V and VIII deficiency
Mild to moderate bleeding
Aka familial multiple clotting factor deficiency Type I
PT/PTT prolonged
disorders of fibrinolytic protein inhibitors
a2-antiplasmin (AP) and plasminogen activator inhibitor-1 (PAI-1)
Impaired regulation of fibrinolysis
Excess plasmin activity
Specific assays for AP and PAI-1
Screening test: PT/PTT, specific factors are all normal, FIB can be decreased
Results in bleeding symptoms
what does thrombin do?
Coagulant function (fibrin formation)
Anticoagulant fxn
Cytokine-like activity/inflammation
Wound healing/cellular proliferation
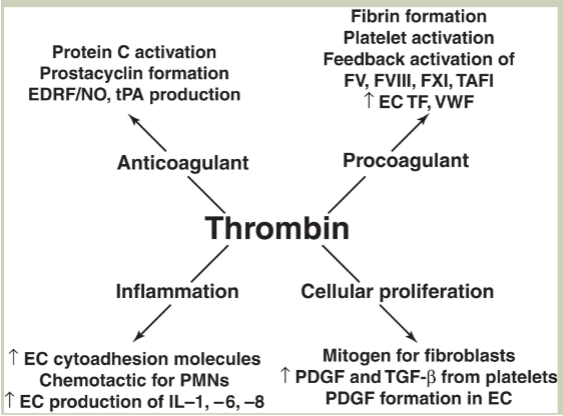
disseminated intravascular coagulation (DIC)
Patient generally bleeds at same time that disseminated clotting is occurring
Results in the uncontrolled inappropriate formation and lysis of fibrin within the blood vessels
Clotting protein, inhibitors, and platelets CONSUMED
Consumed faster than they are synthesized (consumption coagulopathy)
Acquired deficiency of multiple hemostatic components
Fibrinolysis follows fibrin formation
(DIC) incidence and etiology
1 in 1000 hospital patients
Most involved introduction of TF into vascular system
Most common trigger—severe infections (septicemia)
Bacterial toxins and inflammatory cytokines (IL-1, IL-6, TNF) activate EC to express TF
Other common triggers
Pregnancy (thromboplastin from amniotic fluid)
Massive tissue or blood cell injury
Malignancy (increased TF in the blood stream
(DIC) pathophysiology
Initiating event
Generalized or systemic (NOT localized) UNREGULATED formation of thrombin
Consumption of its substrates
Fibrinogen, FV, FVIII, FXIII
Depletion of prothrombin, activation and aggregation of platelets
Thrombin
Activates EC release of t-PA
Activating plasminogen → plasmin
Triggering aggressive secondary fibrinolysis
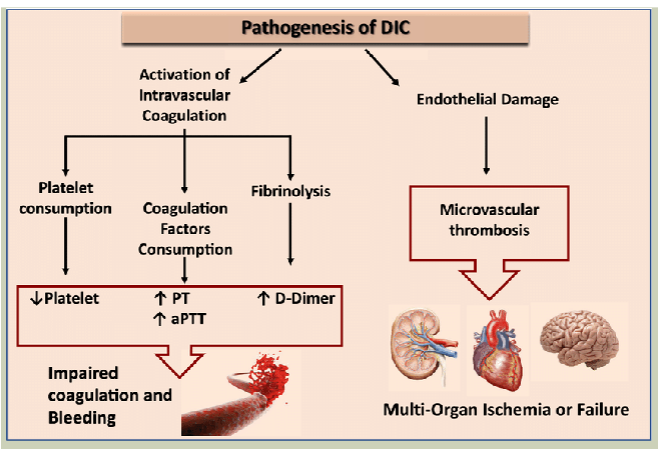
why does DIC occur?
results from failure of mechanisms that limit blood clotting and thrombin generation
AT, HCII, & TM are overwhelmed
IL-1, TNF lead to decrease of endothelial cell expression of TM
Decreased PC/PS inhibitory system
Increased FPA/FPB
Increased D-dimer
symptoms of DIC
DIC can be acute (hemorrhagic) or chronic (thrombosis)
Clinical symptoms
Bleeding from multiple sites ; clots ; bruising
SOB ; confusion
Fibrin strands within small vessels result in traumatic destruction of RBCs (schistocytes)
Microangiopathic hemolytic anemia (MAHA)
Fibrin deposition in an obstruction of microvasculature
Tissue anoxia/microinfarcts
Renal failure, liver failure, respiratory failure, skin necrosis, gangrene, thromboembolism
lab diagnosis of DIC
Available tests are nonspecific
No single or combination of tests can establish the definitive diagnosis of DIC
PT, APTT, and TT—prolonged
↓ fibrinogen & ↓ AT
D-dimer + (not specific for DIC)
Platelet count ↓; platelet function tests abnormal
Schistocytes, thrombocytopenia on PB smear
Markers of ↑ coagulation (thrombin generation)
Not routinely available
therapy for DIC
Eliminate underlying cause if possible
Acute DIC often self-limited & will disappear when fibrin is lysed
Replacement therapy
Platelets, RBCs, cryoprecipitate, or FFP
LMW heparin used when:
Predominant thromboembolic manifestation
Replacement therapy fails to alleviate excessive bleeding
Mortality is still high at 50-60%--shock is common
primary fibrinogenolysis
Must be differentiated from DIC
Plasminogen inappropriately activated to plasmin without concomitant thrombin generation
Plasmin lysis of fibrinogen, not fibrin
Circulating plasmin will degrade
Fibrinogen V, VIII, XIII, other coag factors and plasma proteins
Relatively rare; often seen w prostate disorders, liver dx
Platelet count and D dimer usually normal (no fibrin formation)
Therapy is EACA--epsilon aminocaproic acid (amicar)
Specific inhibitor of plasmin (dangerous if given to pts w DIC!)
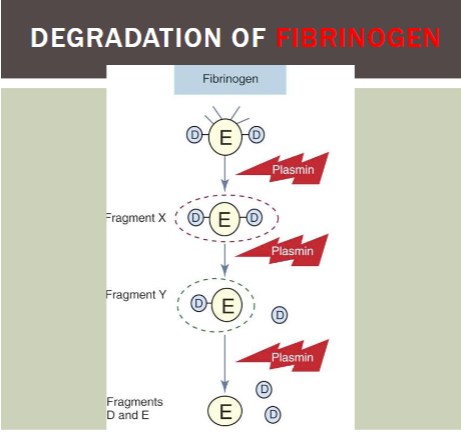
products of FIBRINOGEN cleavage
Fragment X (trinodular)
Fragment Y (dinodular)
Fragment D and E (unimodular)
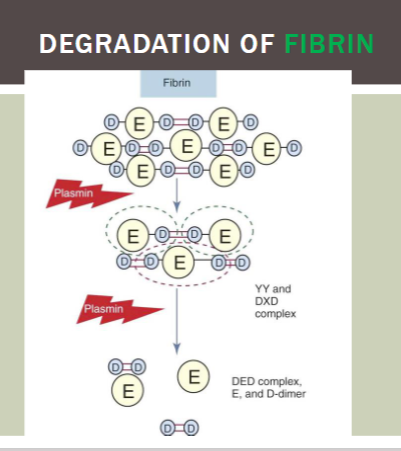
products of FIBRIN cleavage
DD/E
YD/DY
YY/DXD (most common)
DD (most common)
liver’s role in homeostasis
Makes majority coagulation factors
Makes fibrinolytic proteins
Makes inhibitors to coagulation
Clears activated factors
liver damage & bleeding disorders
Affects all hemostatic functions
Liver synthesizes procoagulant & fibrinolytic proteins
Liver macrophages also remove activated factors
Laboratory test results may resemble DIC
Decreased production of all proteins involved in fibrin formation
May cause all screening coagulation tests to be prolonged
Platelet count +/– decreased
+ hypersplenism, ↓ TPO
FDPs ↑
Lack of hepatic clearance of FDPs and plasminogen activators
what does vitamin K do?
Fat soluble vitamin
Functions as a co-factor for carboxylase (activates certain factors)
2, 7, 9, 10, PS, PC, PZ
Essential for Ca2+ binding
structure of coagulation proteins
Catalytic domain
Cleaves peptide
Converts inactive proenzyme to active enzyme
Non-catalytic domains
Regulatory segments
Bind Ca2+ and promote interaction with PL, cofactors, receptors and substrates
vitamin K deficiency
Proteins are not gamma-carboxylated
Ca2+ binding sites are nonfunctional
Called des-y-carboxy-proteins
Induced functional deficiencies of all vitamin K-dependent proteins
Similar to coumadin
Causes of vitamin K deficiency in adults
Malabsorptive syndromes
Biliary tract obstruction
Prolonged broad-spectrum antibiotics--abolishes normal flora
how to differentiate liver disease from vitamin K deficiency in hemostasis results?
factor V remains in normal levels in vitamin K deficiency
vitamin K deficiency in newborns
Vitamin K deficiency bleeding (VKDB)
Newborn hepatic immaturity
Associated with vitamin K-dependent factors at levels of 30–50% of normal adult levels
Bleeding from skin, mucosal surfaces, circumcision site, ecchymoses, large intramuscular hemorrhages
Suspected when PT or APTT more prolonged than expected for age group
(vit K deficiency) bleeding diagnosis + prevention
Specific factor assays for II, VII, IX, and X--all markedly decreased
Platelet count and platelet function tests--normal
Vitamin K administered to all newborns in US standard practice
types of acquired pathologic inhibitors
Usually IgG or rarely IgM immunoglobulins
Inhibitors of single factors
Patients with inherited factor deficiencies
After treatment with replacement concentrates
Associated w diseases, drugs, pregnancy
Interfere with or neutralize clotting factor activity
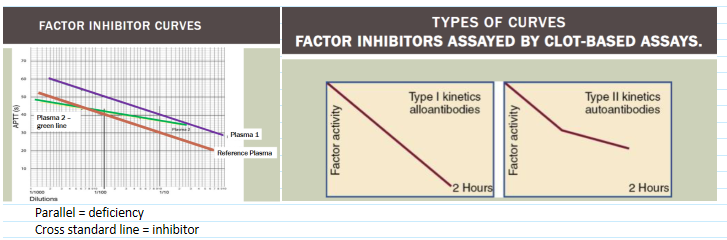
Prolonged screening test not corrected by 1:1 mixture with normal plasma
Or correct initially but not after incubation 37C for 1-2 hours
Anti-phospholipid antibodies/lupus or CIRC anticoagulant
who are the most common factor inhibitors?
VIII and IX
(most common inhibitors) inhibitors to factors VIII + IX
FVIII low responders
Low titer antibodies that do not rise after further exposure to FVIII—give large doses to overwhelm antibody
FVIII high responders
Inhibitors markedly rise foll owing further exposure
give FIX complex products to bypass need for FVIII
aka FEIBA (factor eight inhibitor bypassing activity)
therapy for factor inhibitors
Prothrombin complex concentrates (PCC) or Recombinant FVIIa
Bypass agent that activates FX
Equally effective for FVI II and FI X inhibitors
(factor inhibitors) lupus anticoagulant
Develop in 6-16% of patients w SLE
Also seen in other autoimmune diseases, neoplasias, certain infections, certain drugs, and apparently normal individuals
Interact with phospholipid surfaces of reagents used in APTT and sometimes PT
"Antiphospholipid antibodies" (APLs)
Usually discovered when APTT is unexpectedly prolonged
Laboratory phenomenon
Not associated w clinical bleeding, rather more often associated w thrombosis