Lecture 5 - protein structure
1/77
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
78 Terms
what are peptides and proteins?
Polymers of amino acids
That means many amino acids are linked in a chain.
These chains are called polypeptides or polyaminoacids.
what is a peptide bond?
A peptide bond forms between the carboxyl group (-COO⁻) of one amino acid and the amino group (NH₃⁺) of another.
Peptide bond is the special name given to the amide
bond that joins two amino acids
how is a peptide bond formed?
through condensation
H₂O (water) is removed.
The carbon (C) of the carboxyl group forms a bond with the nitrogen (N) of the amino group, creating a C–N amide linkage (highlighted in blue).
Resulting Molecule
The resulting structure is a dipeptide (a molecule with two amino acids linked).
The highlighted part is the peptide bond (C=O–NH).
what type of geometry are proteins
Proteins are linear chains made by linking amino acids via peptide (amide) bonds.
These bonds are formed by removing water (H₂O) between:
A carboxyl group (
–COO⁻)And an amino group (
–NH₃⁺)
This is a condensation reaction, same as shown before.
what is an example of a simple peptide?
An example of a simple peptide is aspartame
Aspartame is a real molecule made from two amino acids:
Aspartic acid and Phenylalanine methyl ester
Fun fact: It’s used as an artificial sweetener—200× sweeter than sucrose (table sugar)!
what does structure of protein sequence consist of (3)?
Backbone
Side chains
Termini (n -terminus and c-terminus)
what is the backbone of a protein
The backbone of a protein is a repeating pattern of:
–NH–CH(R)–CO– unitsThis backbone is the same in all proteins, regardless of the specific amino acid.
It’s made of amide (peptide) bonds connecting each amino acid.
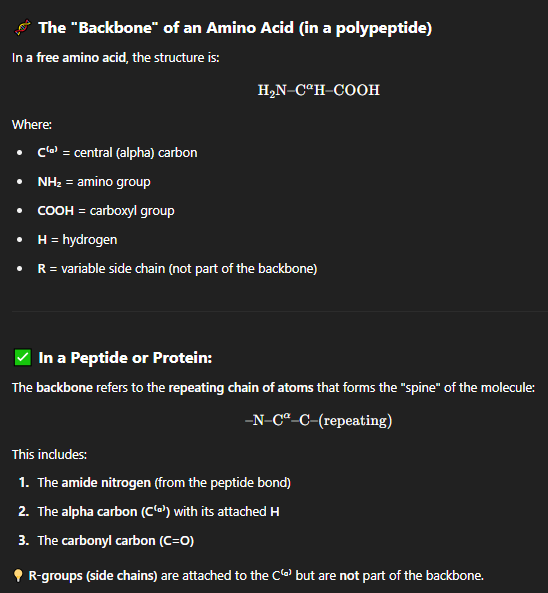
what are sidechains of a protein?
These are the unique parts of each amino acid.
In the diagram:
CH₃ → Alanine
CH₂OH → Serine
(CH₂)₄–NH₃⁺ → Lysine
(CH₂)₂–COO⁻ → Glutamate
Side chains determine the chemical properties of the amino acids (like polarity, charge, size).
what are Termini:?
Amino terminus (N-terminus):
→ The start of the polypeptide chain; has a free –NH₃⁺ groupCarboxy terminus (C-terminus):
→ The end of the chain; has a free –COO⁻ group
characteristics of the three structures?

what happens to the a-amino and a-carboxyl group when a peptide bond forms?
Become covalently bonded into the peptide backbone.
As a result, they lose their ability to ionize in solution—they’re no longer charged.
where are charged group present in polypeptides?
The only charged parts in a polypeptide:
The two termini:
→ N-terminus (can still carry a + charge)
→ C-terminus (can still carry a – charge)Some R groups (side chains) depending on the amino acid (e.g., Lys, Arg, Glu, Asp).
what are residues?
residues refer to the individual amino acids that are part of a peptide or protein chain.
During peptide bond formation, each amino acid loses a water molecule (through a condensation reaction).
So what's left behind in the chain is only a "residue" of the full amino acid — the part that remains after losing H₂O.
A residue is just an amino acid unit within a peptide or protein, after it's been joined by a peptide bond and lost a water molecule
How We Write Protein Sequences?
Always write from N-terminal (left) to C-terminal (right).
Example format:
→ NH₃⁺–AA₁–AA₂–...–AA₆–COO⁻
(AA = amino acid residues)
what is an important structural characteristic of polypeptide chains?
Polypeptides (chains of amino acids) are not rigid—they can bend and rotate at many points.
Their 3D shape (conformation) is essential for their function.
Think of the chain like a bendable wire that must fold into a specific shape to "work."
how is folding predictable of a polypeptide?
Protein folding is driven by the amino acid sequence itself (called the "primary structure").
We now know how to predict protein structure:
Thanks to large datasets from X-ray crystallography and NMR spectroscopy.
And computational tools like AlphaFold (implied here though not mentioned directly).
what are the four levels of protein structure?
primary
seconday
tertiary
quaternary
what is the primary structure?
This is the linear sequence of amino acids in a polypeptide.
It's like spelling a word with letters—each amino acid is one letter.
The order is critical: even one change (a mutation) can drastically affect the protein’s function.
what is the secondary structure
These are local folding patterns within the chain, such as:
α-helices
β-sheets (or β-strands)
These patterns are stabilized by hydrogen bonds between the backbone atoms (not side chains).
what is the tertiary structure?
This is the 3D shape of the entire polypeptide chain.
It’s formed by:
Interactions between side chains (R groups):
Hydrophobic interactions
Hydrogen bonds
Ionic bonds
Disulfide bridges (covalent)
Tertiary structure determines the function of the protein.
what is the quaternary structure
Applies only when there are multiple polypeptide chains (subunits).
It refers to how these subunits interact and arrange to form the final functional protein.
Examples: Hemoglobin (4 subunits), DNA polymerase.
Why This Matters:
Structure determines function—misfolding can cause diseases (e.g., Alzheimer’s, prions).
Understanding structure helps with drug design, enzyme engineering, and biotech.
what was The Protein Folding Problem?
The challenge: Can we predict a protein’s 3D structure from just its amino acid sequence?
This was long considered one of biology’s "grand challenges", as small changes in sequence can cause huge structural shifts.
what was the solution to this problem?
The Solution: AlphaFold
The study cited (Senior et al., Nature, 2020) refers to AlphaFold, an AI model by DeepMind.
AlphaFold uses deep learning to accurately predict protein structures.
It matches experimental results with extremely high precision, often with >90 GDT (Global Distance Test) scores.
Why This Matters: ?
Rapid structure prediction can accelerate:
Drug discovery
Understanding disease-causing mutations
Enzyme design in biotechnology
who discovered the first protein sequence?
The first protein sequence was determined in the
early 1950s by Fred Sanger (Nobel Prize, 1958)
This success was followed by the direct sequencing of thousands of proteins
He later also developed a method for DNA sequencing and won another Nobel in 1980. That method is now automated and used worldwide.
How Sequences Are Determined Today:?
We typically don’t sequence the protein directly anymore.
Instead, we:
Sequence the gene (DNA)
Use the genetic code to translate that into the amino acid sequence
what is Sanger protein sequencing?
which was the first method used to determine the amino acid sequence of a protein.
what are the Sanger Protein Sequencing Steps?
You label only the first amino acid using FDNB (Sanger's reagent).
Then you completely hydrolyze the peptide — breaking all peptide bonds.
So you destroy the entire peptide, and only the original N-terminal amino acid can be identified (as a DNP-amino acid).
The rest of the amino acids are unlabeled and mixed up — you can’t sequence further from the same peptide.
“Sanger sequencing only gives you one amino acid unless you break the protein into smaller fragments, analyze each, and rebuild the sequence using overlaps from different cuts.”
problems with sangers protein sequence method?
what if you want to go beyond the first amino acid with sanger method, what must you do?
Only tells you the first amino acid (N-terminal). also destroys the entire peptide chain
To sequence the entire protein, it must be broken into smaller peptides and analyzed stepwise.
to go further you must
Use Different Proteases Use specific enzymes (proteases) that cut at known amino acid residues:
Sequence Each Fragment
Use Sanger’s reagent (FDNB) on each fragment to label and identify the N-terminal residue.
Fully hydrolyze the fragment to get the DNP–amino acid (N-terminal identifier).
Analyze the amino acid content of each fragment (manually or chemically).
Reconstruct the Full Sequence Using Overlaps
Look for overlapping regions between fragments from different enzyme digests.
These shared sequences act like puzzle pieces that help determine the order of fragments.
what are used to help fragment proteins?
For proteins, sequence specific proteinases
(enzymes that cleave other proteins) and a
few chemical reactions are used
Trypsin
Chymotrypsin
CNBr
what does trypsin do?
cleaves after Lys or Arg (basic residues
what does Chymotrypsin do?
cleaves after Phe, Tyr or Trp (aromatic residues)
what does CNBr (cyanogen bromide) do?
cleaves after Met residue
how is sequencing of fragments of peptides done?
by using the Edman degradation method—one of the foundational techniques for protein sequencing.
how does edman degradation method work?
Chemical Used: Phenylisothiocyanate (PITC)
Reacts with the N-terminal amino acid of a peptide.
Step-by-Step Process: of edman degradation method (5)
Phenylisothiocyanate (PITC) reacts with the free amino group of the N-terminal residue.
Under acidic conditions, the first amino acid is cleaved off (without breaking the rest of the chain).
That amino acid becomes a stable, identifiable de
rivative (shown as a cyclic compound).
The rest of the peptide remains intact and can be cycled through again.
The derivatized residue is analyzed—typically by chromatography—to identify it.

This process is repeated step-by-step, revealing one amino acid at a time.
accuracy of edman degradation?
In ideal cases, this can go up to >80 residues, but realistically, it’s more reliable up to ~30 residues due to loss of accuracy over cycles.
The process is now automated, making it faster and more efficient.
what is an alternative method for determing a protein’s primary structure?
mass spectrometry (MS)
What is Mass Spectrometry (MS)?
A powerful analytical technique used to measure the mass of molecules with extreme precision.
Especially useful for identifying peptides and proteins after enzymatic digestion.
General Workflow: of MS
The standard approach is to first digest our protein
with a protease (like trypsin!), and then identify the
peptides based on their mass
The 4 Key Steps of Mass Spectrometry:
Generation of ions
– The peptides are ionized (e.g., by electrospray ionization or MALDI).Acceleration
– Ions are accelerated using electric or magnetic fields.Separation
– Ions are separated based on their mass-to-charge ratio (m/z) in a mass analyzer.Detection
– A detector measures the arrival time and abundance of each ion to generate a mass spectrum.
Step-by-Step Breakdown: of MS
Ionization & Vacuum Injection
Peptides are dissolved in solution and sprayed into a vacuum chamber through a fine glass capillary.
A high voltage is applied to ionize the sample—this gives the molecules an electric charge.
2. Flight & Detection
The ions are pulled through a vacuum interface and into the mass spectrometer.
Here, they’re accelerated and separated based on mass-to-charge ratio (m/z).
what is the “Time of Flight” Principle:
Lighter ions fly faster and hit the detector sooner.
Heavier ions fly slower and arrive later.
The flight time helps calculate their mass very precisely.
these methods so far
Sanger sequencing (with fragmentation and Edman degradation),
Mass spectrometry (MS),
Protease cleavage patterns,
are used to find what?
are tools to determine the primary structure of a protein — that is, the exact linear sequence of amino acids.
where does the power of mass spectrometry come from?
Thanks to genome projects, we now know nearly all genes and their DNA sequences in organisms like humans and mice.
Since genes encode proteins, we can predict all possible protein sequences in that organism’s proteome.
what is a mass fingerprint?
Every protein, when digested with trypsin, produces a unique pattern of peptide fragments (called a mass fingerprint).
Mass spectrometry measures the mass of those peptides.
what do you get when you compare the masses?
By comparing the measured masses to a database of predicted tryptic digests, we can quickly match the peptide set to a known protein.
This is the core of:?
Proteomics = the large-scale study of all proteins in a cell, tissue, or organism.
Focuses on identifying and quantifying proteins.
Now largely automated with software and databases (like Mascot, SEQUEST).
What is Secondary Structure?
It’s the local, repetitive folding patterns of the polypeptide backbone, not the side chains.
These patterns are stabilized by hydrogen bonds between:
The carbonyl group (C=O) of one amino acid, and
The amide hydrogen (N–H) of another nearby amino acid.
The carbonyl oxygen acts as the hydrogen bond acceptor
The amide hydrogen acts as the hydrogen bond donor
Two main types of secondary structure?
α-Helix
β-Conformation (β-sheet)
what is the α-Helix (3)
Defined by Linus Pauling, the α-helix is:
A right-handed spiral shape.
Hydrogen bonds form every 4th residue.
R groups stick outward from the helix.
what is the β-Conformation (β-sheet) (3)
Polypeptide chains lie side-by-side, forming sheet-like structures.
Hydrogen bonds form between strands, either parallel or antiparallel.
R groups alternate above and below the plane of the sheet.
features of the a-helix structure (5)
The Peptide Bond is Planar
R groups alternate on opposite sides of the peptide backbone—this is called the trans configuration.
Hydrogen bonds form between every 4th amino acid
In an α-helix, side chains (R groups) project away from the helix axis.
α-Helix Has a Dipole Moment
what is a planar peptide bond?
A planar peptide bond refers to the flat, rigid geometry of the bond that links two amino acids in a protein. This occurs because of resonance between the carbonyl (C=O) and amide (C–N) groups.
Why is it planar in alpha helix
The peptide bond (C–N) between amino acids is not a simple single bond.
It has partial double bond character due to resonance between:
the carbonyl group (C=O) and the nitrogen lone pair.
This partial double bond character shortens the bond and makes it less flexible.
consequence of planar peptide bond (3)
The C–N bond is shorter than a regular single bond (1.32 Å).
Rotation is restricted — meaning the bond is rigid and planar. Rotation is allowed around the α-carbon (Cα), but not around the peptide bond (C–N).
This constraint is essential for defining stable 3D structures like α-helices and β-sheets.
what is trans configuration.?
In a stable α-helix, R groups alternate on opposite sides of the peptide backbone—this is called the trans configuration.
The cis configuration (where R groups are on the same side) leads to steric clashes (physical crowding) and is energetically unfavorable.
The α-helix formation requires trans peptide bonds to prevent R group collisions.
Cis peptide bonds = R group clashes = unstable
Trans peptide bonds = R groups far apart = stable, preferred
Why does trans configuration matter?
These structural constraints are universal rules for peptide chains in proteins.
They help explain why α-helices form the way they do, and why some residues (like Proline) disrupt them (more on that later).
why do R groups point outwards? (2)
In an α-helix, side chains (R groups) project away from the helix axis.
This minimizes steric clash and allows the helix to pack closely with other structures.
what three types of residues disrupt alpha helix formation
If you have a cluster of:
Charged residues → electrostatic repulsion
Bulky residues (e.g., Trp, Phe) → steric hindrance
Small, flexible residues (e.g., Gly, Ala, Ser) → instability
These can disrupt helix formation.
how does α-Helix Have a Dipole Moment?
The alignment of many polar peptide bonds (each with a C=O and N–H) in the same orientation gives the helix an overall dipole:
Partial positive (+) at N-terminus
Partial negative (–) at C-terminus
The α-helix has a dipole moment because:
Every peptide bond has a dipole
These dipoles all point in the same direction in the helix
They add together, creating a net helix dipole from N to C terminus
what are the affects of this dipole moment?
This dipole can affect how the helix interacts with other molecules:
Positively charged N-terminal end can attract negative ions or phosphate groups
Negatively charged C-terminal end can attract positive charges (e.g., metal ions)
Helps with protein folding, stability, and ligand binding
how do hydrogen bonds form in alpha helix?
Due to the resonance of the peptide bond:
The nitrogen (N–H) has a partial positive charge (δ⁺)
The carbonyl oxygen (C=O) has a partial negative charge (δ⁻)
This charge separation allows the H-bond to form:
when do hydrogen bonds form in alpha helix?
In an α-helix, a hydrogen bond forms between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4.
This repeated pattern stabilizes the helical turn.
What does "i" and "i+4" mean?
"i" refers to an amino acid at a particular position in the sequence.
"i+4" means the amino acid that is four residues ahead of "i" in the linear sequence.
So, if:
"i" = amino acid #1
Then "i+4" = amino acid #5
why do hydrogen bonds matter in alpha helix structure? (3)
These internal H-bonds:
Stabilize the helical structure
Allow tight and consistent right-handed coiling
Are directional—pointing from N-terminus to C-terminus
Features that disrupt A-helix (4)
a) Electrostatic repulsion
b) Bulky R-groups such as isoleucine
c) Small residues such as Gly, Ser & Ala favor conversion to the B-conformation
D) Proline disrupts the helix
how does Electrostatic Repulsion disrupt alpha helix?
Clustering of Like-Charged Side Chains (R groups)
If multiple charged amino acids (like Lys⁺, Arg⁺, Glu⁻, Asp⁻) are located near each other in the helix,
And they have the same charge (e.g., several Lys⁺),
Their side chains repel each other.
Result:
This repulsion causes strain on the helical backbone
May bend, twist, or even break the helix
Reduces overall stability and likelihood of helix formation
Note* - In an α-helix, side chains stick out and down from the helical backbone. If similarly charged side chains are spaced closely (e.g. every 3–4 residues), they end up on the same face of the helix — close enough to repel each other.
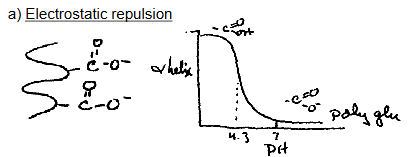
what does this graph describe?
The graph shows α-helix formation of polyglutamate as a function of pH.
At high pH (~>4.3), glutamate side chains are deprotonated (–COO⁻) → strong repulsion → helix unfolds.
At low pH (<4.3), glutamates are protonated (–COOH) → neutral → helix forms.
key characteristics of electrostatic interactions
Too many like charges (all + or all –) in a helix = repulsion = disruption.
However, favorable electrostatic interactions (oppositely charged residues) can also stabilize helices (e.g., Glu⁻ with Lys⁺).
how do Bulky R-Groups disrupt alpha helix?
Bulky side chains like those of Ile, Leu, Val, Phe, Trp can physically clash with other nearby R groups when packed in an α-helix.
Bottom line: Too many bulky residues in close proximity destabilize the helix by creating steric interference.
how do Small Residues (Gly, Ser, Ala) disrupt alpha helix?
These residues are too flexible or lack sufficient steric bulk to support the rigid structure of an α-helix:
Glycine is overly flexible (no side chain at all).
Serine has a small polar side chain (–CH₂OH).
Alanine, although often helix-friendly, in high concentration can also lead to β-sheet formation.
Bottom line: Small, overly flexible side chains may promote β-sheet (β-conformation) over α-helix, especially when present in clusters.
steric bulk = refers to the physical size and spatial demand of atoms or groups in a molecule
what is helical penalty?
Helical penalty is a measure of how much an amino acid resists forming an α-helix.
It tells you how favorable or unfavorable it is for a particular amino acid to be part of an α-helix based on:
Sterics (size and shape of R group)
Hydrogen bonding ability
Backbone flexibility or rigidity
how is it measured?
The penalty is usually given in kcal/mol.
A higher number means greater energetic cost to include that amino acid in a helix (i.e., it disrupts helix stability).
A lower or negative number means the residue favors helix formation.
key amino acid helical penalties (4)
Key ones highlighted:
Alanine (Ala):
Low penalty (0.00) → one of the most helix-favorable residues.
Proline (Pro):
Very high penalty (3.26 kcal/mol) → strongly disrupts α-helices.
Glycine (Gly):
Moderate penalty (1.00) → its flexibility makes it less favorable in helices.
Cysteine (Cys):
Moderate disruptor (0.77), possibly due to its ability to form disulfide bonds or steric effects.
Why does Proline Break α-Helices
Proline's side chain forms a ring that locks its nitrogen into a rigid sp³ configuration (tetrahedral), preventing it from becoming planar like a normal peptide bond.
what’s the consequences of this? (3)
Consequences:
The amide nitrogen can't donate a hydrogen → can't form a hydrogen bond.
The rigid ring forces a kink in the helix → introduces a sharp bend.
For this reason, proline is often found at the ends of helices, not in the middle.
summary of proline affects?
Proline is a “helix breaker” because its rigid ring and lack of H-bonding disrupt the regular α-helix structure. Other residues (like Gly or bulky ones) also weaken helices depending on their chemistry or size.