Module 6: Signal Transduction
1/94
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
95 Terms
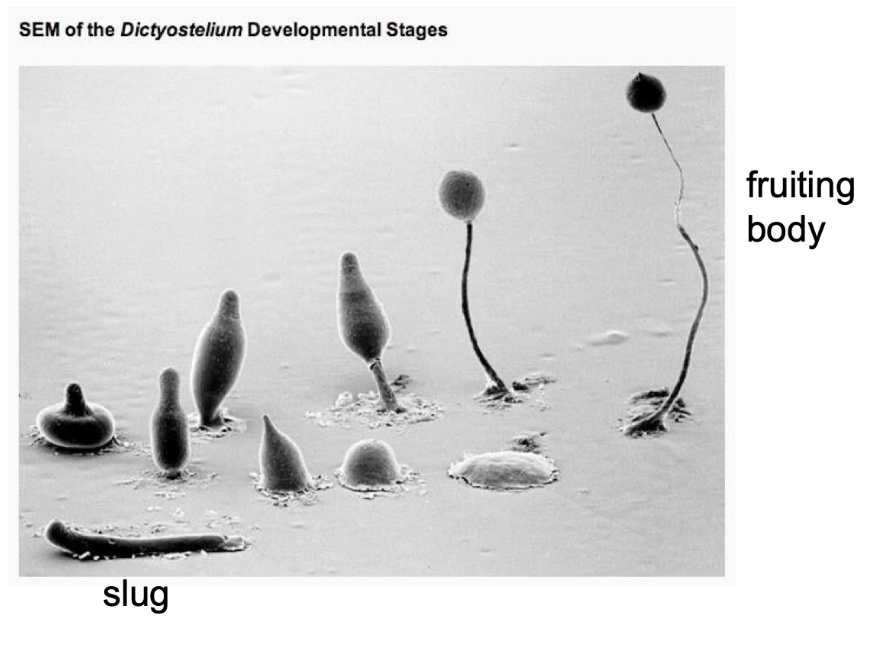
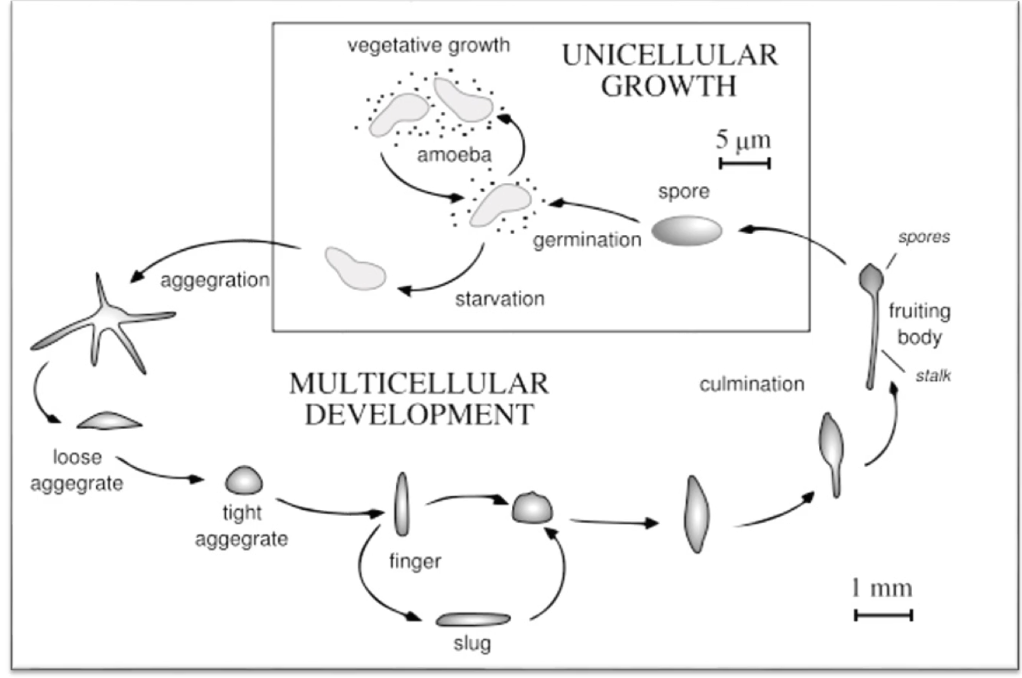
What is Dictyostelium discoideum and how does it respond to resource scarcity?
Eukaryotic slime mold with both unicellular and multicellular stages
Transitions:
From single amoebae to multicellular slug
Then to a fruiting body
During food scarcity:
Cells aggregate to form a slug
Slug migrates toward heat, light, and humidity to find food
Differentiates into:
Anterior → prestalk cells
Posterior → prespore cells
In suitable environment:
Anterior forms stalk
Posterior forms spores of the fruiting body (~2 mm tall)
What triggers aggregation in Dictyostelium and what is the outcome?
Vegetative Growth Phase: Food Abundant
Feed on bacteria (E.coli)
Amoeba divide by mitosis
Trigger: Starvation
Signal: Cyclic AMP (cAMP)
Secreted by starved cells
Attracts other amoebae
Outcome:
Aggregation into slug
In a nutrient rich environment, differentiation into stalk and spores
Spores have hard cell walls (dormant survival)
Spore germination occurs when food returns and new single-celled amoebae form
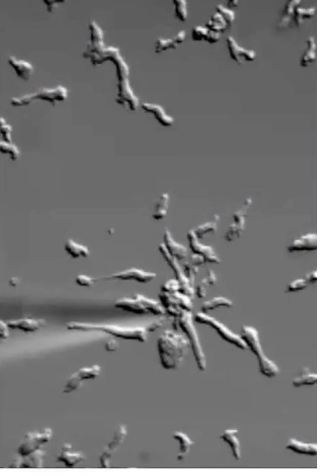
How do Dictyostelium cells detect and respond to cyclic AMP (cAMP) during aggregation?
cAMP acts as aggregation signal
Detected by GPCR which binds cAMP on extracellular domain
Triggers actin cytoskeleton reorganization
Allows cells to move toward cAMP source
Example: cells in dish move toward pipette releasing cAMP
How do Dictyostelium cells physically respond to cAMP?
Extend filopodia toward signal
Undergo actin reorganization:
Nucleation
Polymerization
Depolymerization
Dynamic movement is directed toward cAMP source

How does a clathrin heavy chain mutation affect Dictyostelium’s response to cAMP?
Mutation blocks vesicle formation → no protein transport to membrane
GPCR (receptor for cAMP) can't reach cell surface
cAMP still present (signal source unchanged)
Cells can form filopodia but show no net movement
Reason: no surface GPCR → no cAMP detection → no directed movement
How do neutrophils in humans respond to bacterial infections?
Detect chemical signals from bacteria
Move towards signal source (chemotaxis)
Process:
Surface receptors bind signal
Internal signaling cascade activates
Leads to movement and endocytosis of bacteria
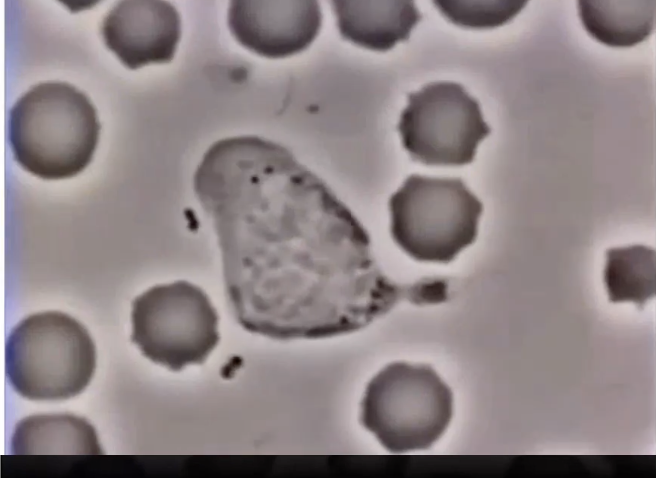
How do neutrophils detect and respond to bacterial signals?
Neutrophils = type of white blood cell
Detect chemical signals from bacteria
Surface receptors bind bacterial signal
Triggers internal signaling cascade
Leads to directed movement toward bacteria (chemotaxis)
Eventually engulf bacteria via endocytosis
What signal do neutrophils detect to find bacteria, and how do they respond?
Bacteria unintentionally release fMLP (formylated-Met-Leu-Phe peptide)
fMLP = bacterial protein fragment
Neutrophils have GPCR on surface that binds fMLP
GPCR activation → intracellular signaling cascade
Neutrophil moves toward fMLP source
What is cell signaling and what are its key steps?
Cell signaling = transmission of info from one cell to another
Must lead to change in behavior in receiving cell
Signal alone is not useful without a response
Key steps:
Production & release of signal
Perception by receptor (usually on cell surface)
Interpretation via intracellular pathways
Cellular response (e.g., movement, division, secretion)
Outcomes of STP
Signal transduction pathway (STP): Series of chemical events inside cell
Leads to changes in target cell behavior, e.g.:
Gene transcription
Cell movement/growth
Cell differentiation
Metabolic changes (enzyme activation/inactivation)
Signal must be removed to stop response
Only cells with the correct receptor can respond to the signal
What determines specificity in signal-receptor interactions?
Based on molecular complementarity
Factors:
Shape and fit of interacting surfaces
Non-covalent interactions (e.g., hydrogen bonds)
Specific amino acid residues in:
Signal molecule
Receptor
Single amino acid changes can disrupt binding
Receptors usually bind only one natural ligand or closely related molecules
Receptor binds signal:
Conformational change in receptors intracellular domain → triggers signal transduction cascade → cellular response
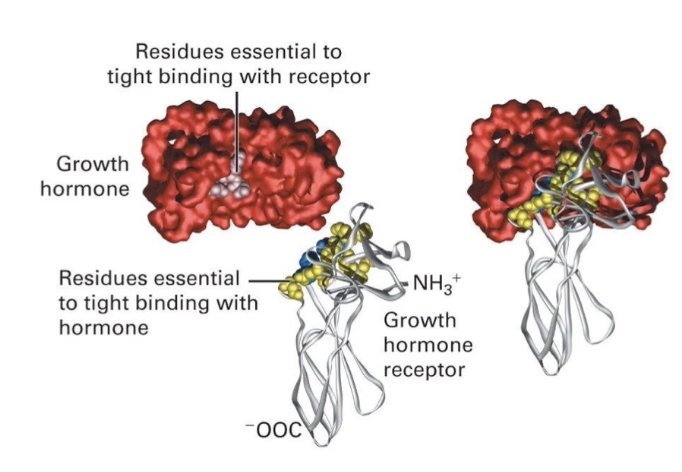
How is specificity of the signal response achieved in cells?
First level: specificity of ligand-receptor binding
Only cells with the matching receptor respond
Second level: specificity of intracellular response
Same signal can activate different proteins in different cells
Leads to varied outcomes (e.g., transcription factor activation, movement, metabolism)
Signal transduction pathway translates extracellular signal into specific cellular response
Specificity determined by internal signal transduction pathway
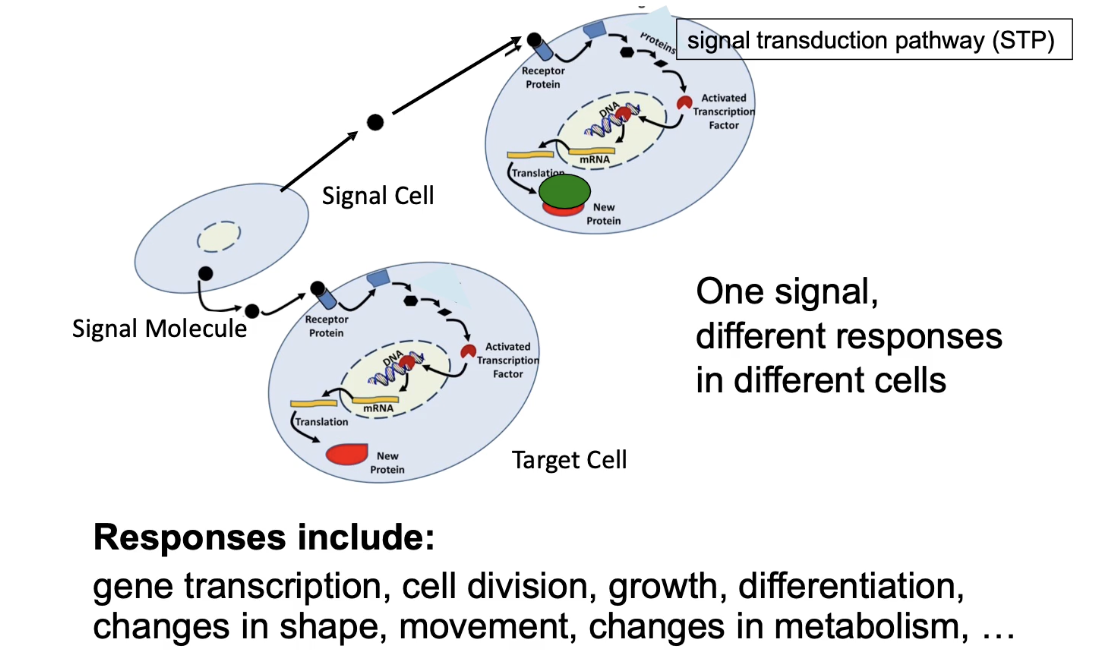
What distinguishes fast and slow cellular responses to signals?
Fast response:
Signal binds membrane receptor
Activates cytosolic enzyme by modification (phosphorylation, methylation, acetylation)
Changes activity of existing proteins
Rapid response
Slow response:
Signal passes through membrane to soluble cytosolic receptor
Receptor moves to nucleus
Acts as transcriptional activator → mRNA produced → protein synthesis
Requires transcription, translation, folding, modifications
Slower response
Same signal & receptor types can trigger either response depending on cell type
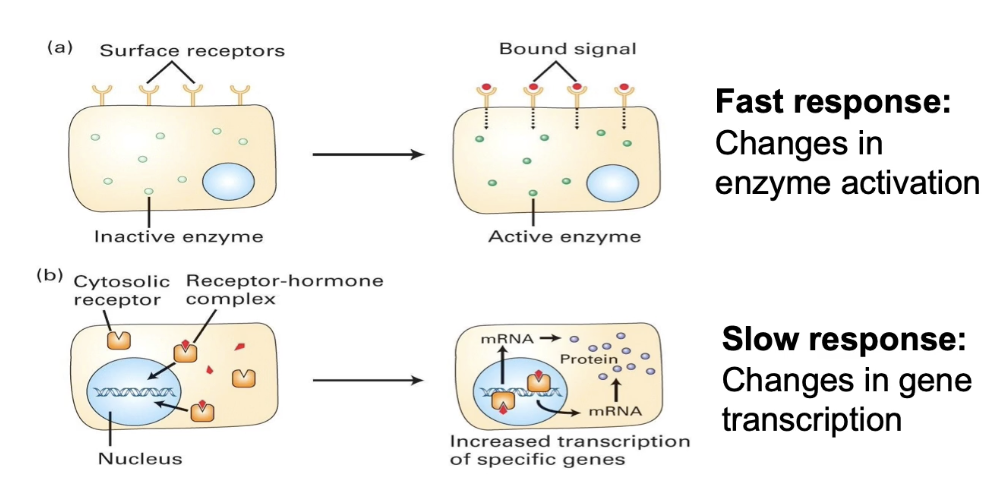
How can receptor-signal binding affinity and cellular physiological response be measured, and what does their comparison reveal?
Measure receptor affinity by plotting ligand concentration vs. fraction of receptors bound
Kd = ligand concentration at half-maximal binding (rep receptor-signal affinity)
Measure physiological response by plotting ligand concentration vs. fraction of cells responding
Half-maximal response concentration often lower than Kd
Indicates signal amplification inside the cell
Small amounts of signal can trigger a large cellular response
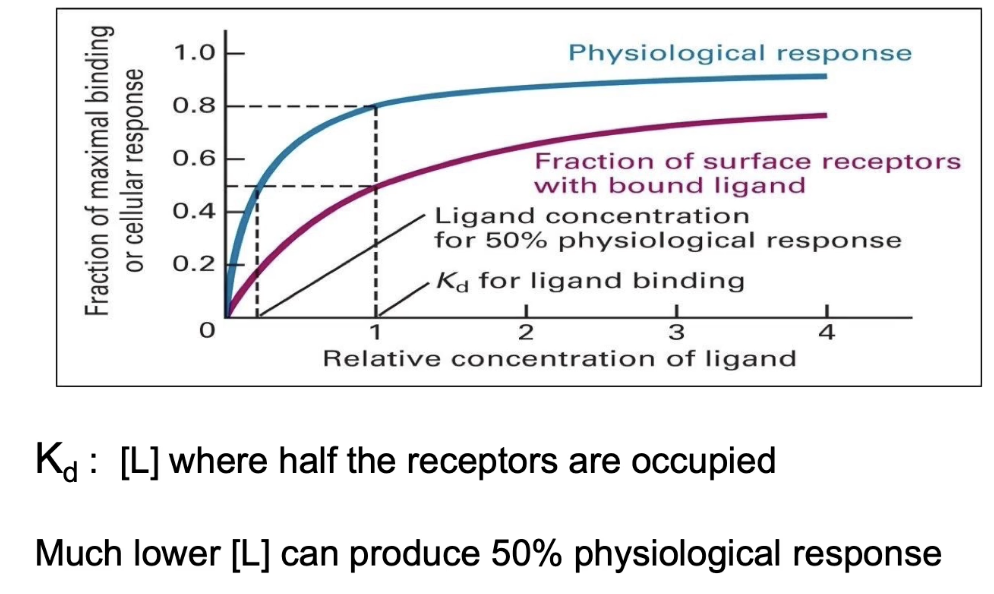
What are the main differences between endocrine and paracrine intercellular signaling?
Endocrine signaling:
Signals (usually hormones) secreted into circulatory system
Signal reaches distant target cells throughout body
Many different tissues can respond simultaneously
Signaling and target cells are far apart
Paracrine signaling:
Signals released into extracellular space
Affect nearby neighboring cells
Common signals: growth factors, neurotransmitters
Signaling and target cells are close together
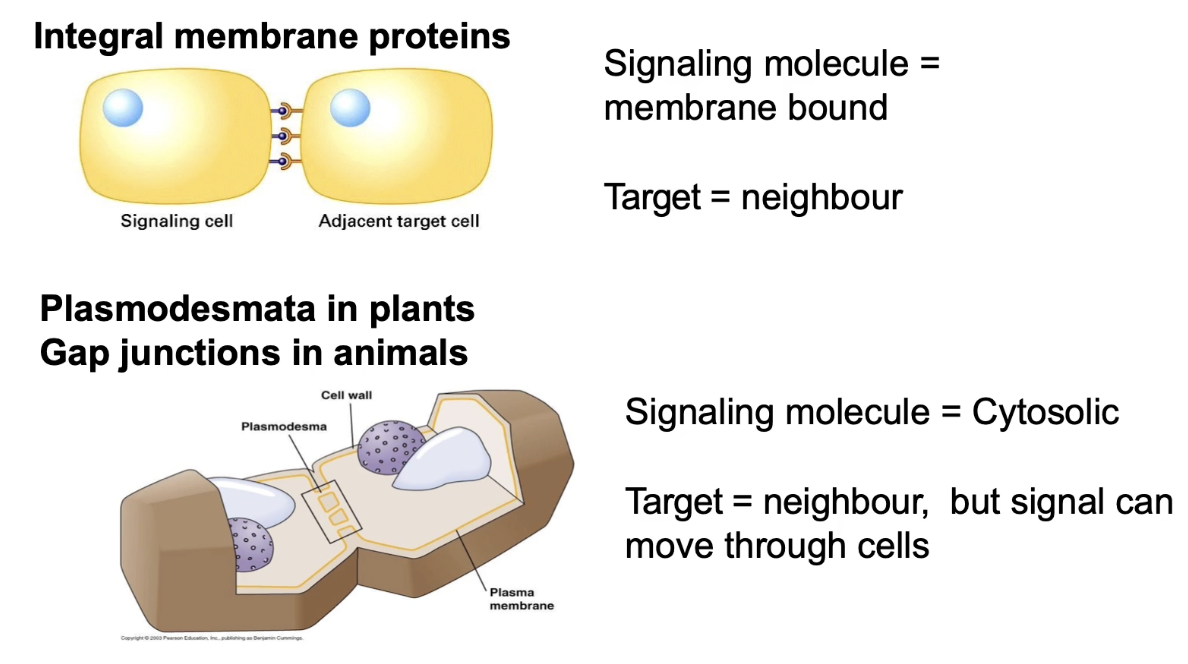
What is proximal signaling and how do cells communicate through direct contact or cytosolic connections?
Proximal signaling: signal and receptor are transmembrane proteins on adjacent cells
Requires direct cell-cell contact via cell integral membrane proteins
Plants: use plasmodesmata — channels connecting cytoplasm across cell walls, allowing fast cytosolic messenger movement
Forms vascular system to transfer signals from root → leaves
Animals: use gap junctions — cytoplasmic channels for diffusion of small molecules/secondary messengers
Allows coordination of responses by sharing internal signals between neighboring cells
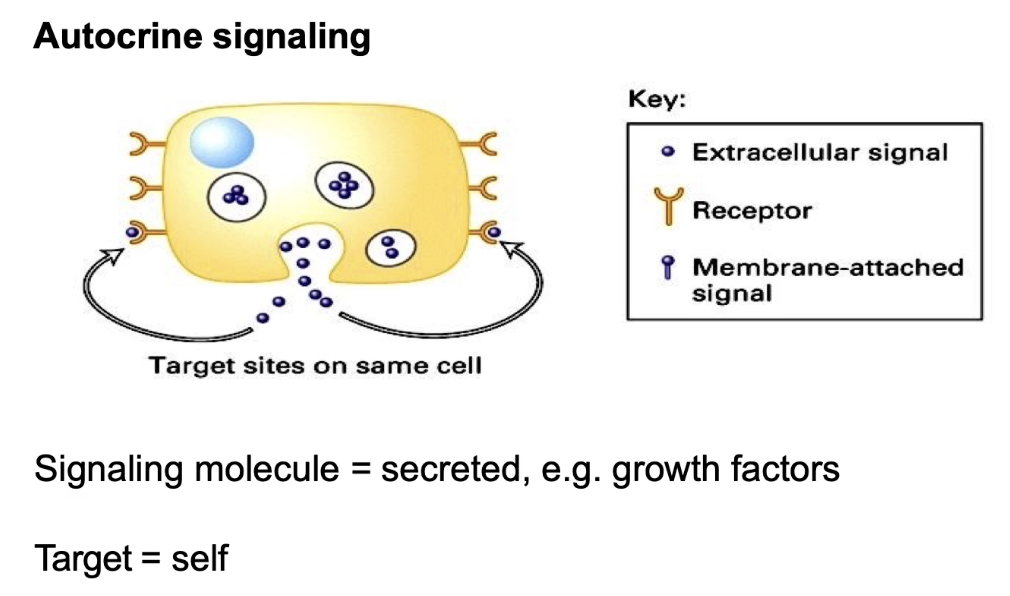
What is autocrine signaling and give an example?
Cell signals to itself (signaling cell = target cell)
Produces and responds to its own secreted signal
Example: growth factors that regulate cell division (promote or inhibit)
Allows a cell to self-regulate based on internal and external conditions
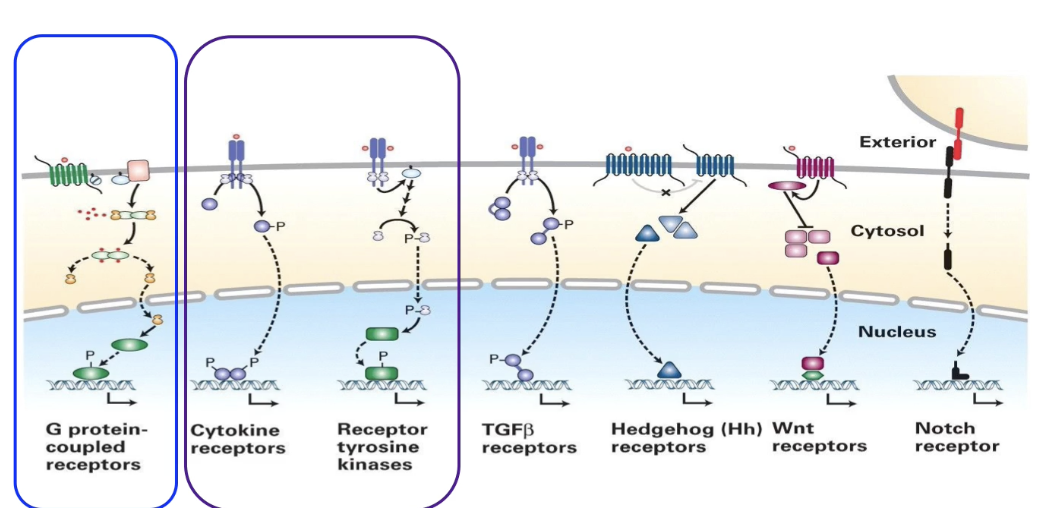
What are the 3/7 major types of cell surface receptors focused on in this module, and what signaling pathways do they involve?
Cytokine receptors: involved in JAK/STAT pathway, regulate red blood cell production
Receptor Tyrosine Kinases (RTKs): linked to phosphorylation cascade via small G-protein Ras, regulate gene expression
G-Protein Coupled Receptors (GPCRs): activate effector proteins to produce second messenger cAMP, regulate cell metabolism
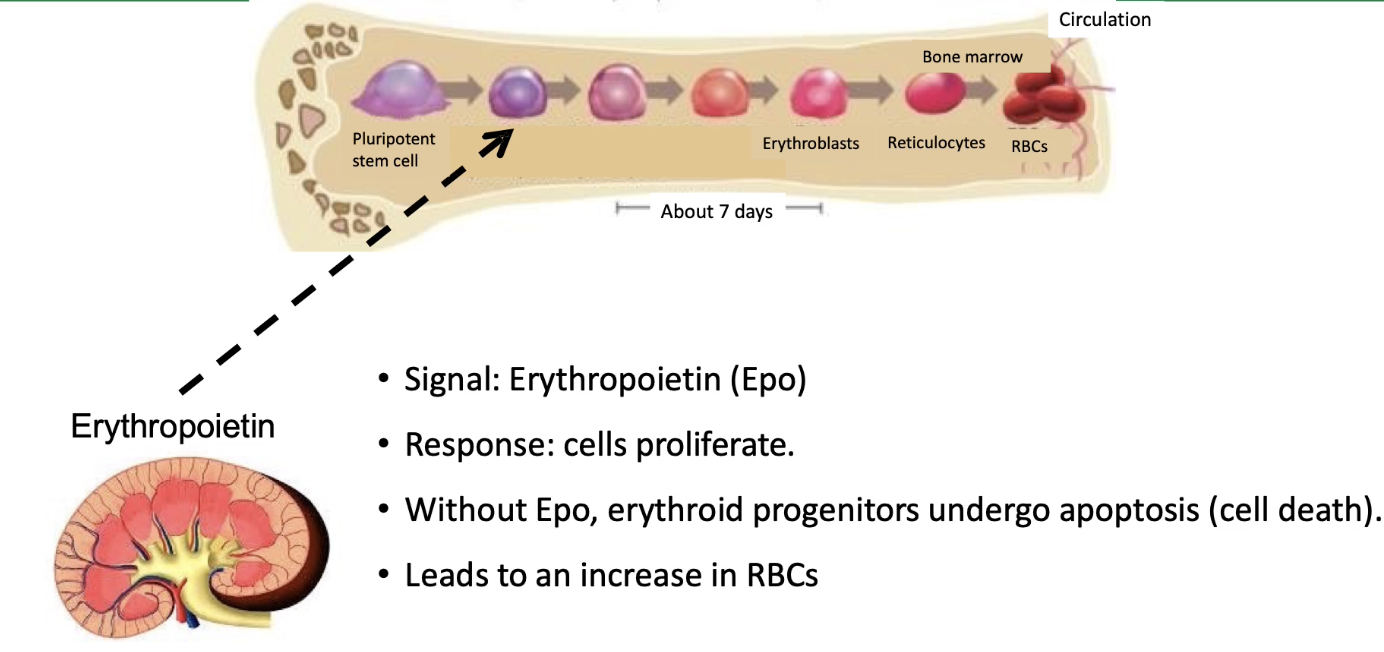
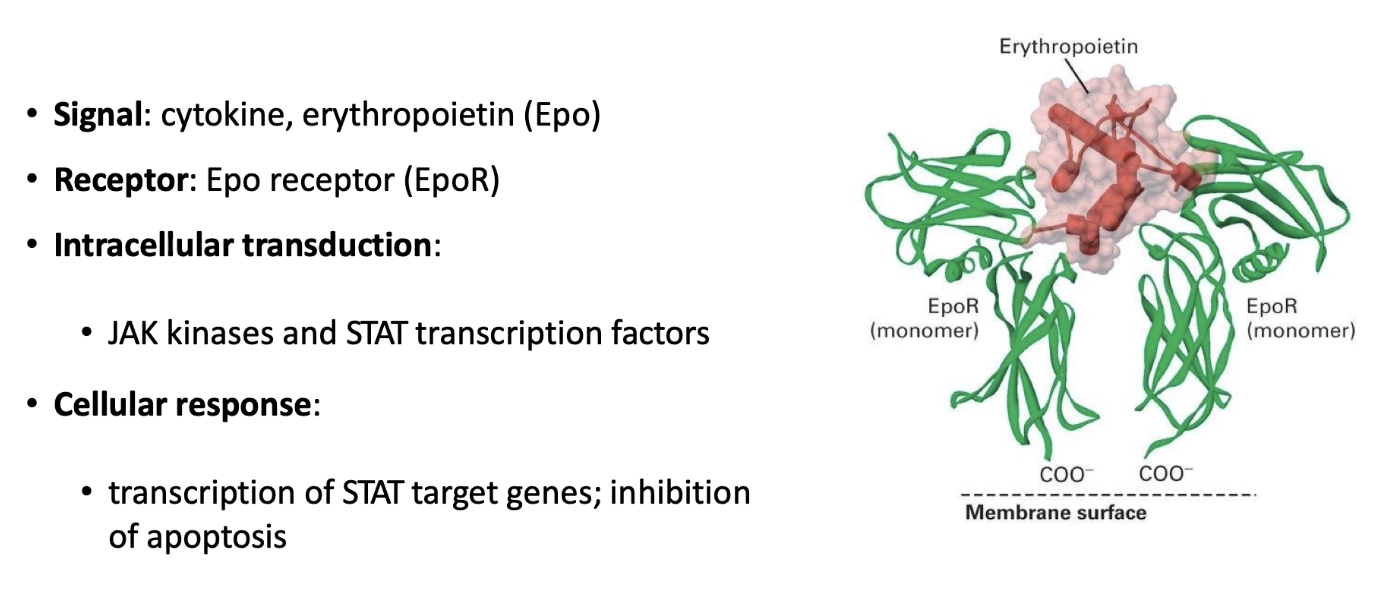
How does erythropoietin (Epo) regulate red blood cell production?
Adult human body produces ~2 million new erythrocytes (red blood cells) per second
Develop in bone marrow, circulate ~4 months, then recycled by macrophages
Red blood cells replaced when pluripotent stem cells (progenitor cells) stop dividing and differentiate
Erythropoietin (Epo) is the key cytokine signal triggering erythrocyte maturation
Epo production regulated by an oxygen-binding transcription factor in kidney cells
Epo is secreted into the circulatory system (public signal)
Only erythrocyte progenitor cells express the erythropoietin receptor (EpoR), a cytokine receptor
Binding of Epo to EpoR activates the JAK-STAT signal transduction pathway
JAK-STAT activation causes:
Inhibition of apoptosis (cell death) in progenitor cells
Changes in gene expression patterns supporting maturation
Commitment of progenitors to differentiate into mature erythrocytes
How is the EpoR activated?
Inactive state:
EpoR is a monomeric, single-pass transmembrane protein, inactive alone
Signal binding:
One Epo molecule binds two EpoR monomers, causing receptor dimerization
This brings cytosolic domains close together, initiating intracellular signaling
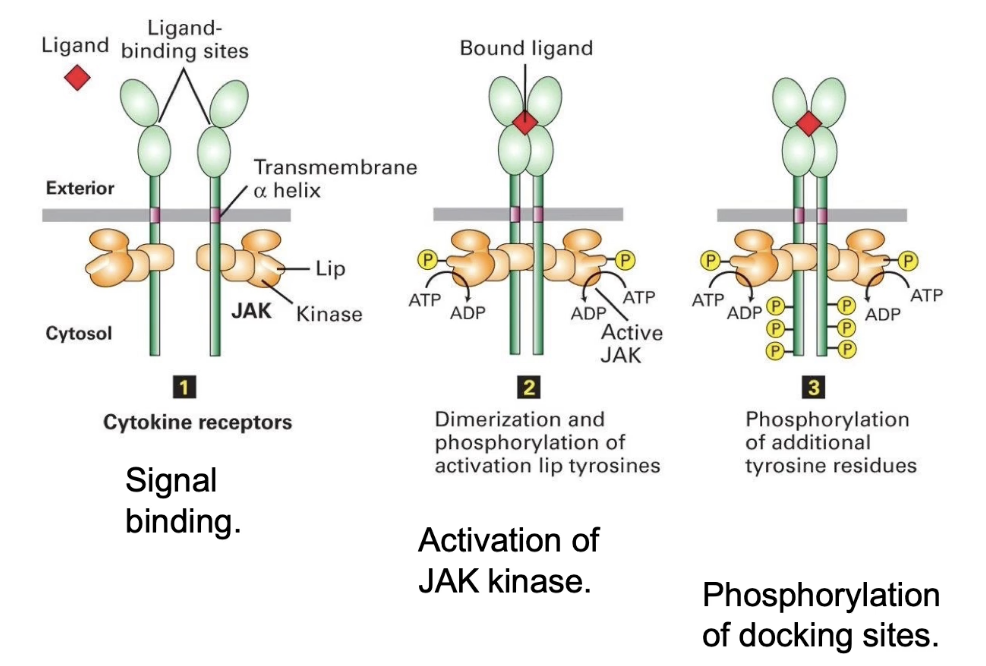
What happens when erythropoietin binds to its receptor?
Epo receptor has 3 domains:
Extracellular domain (binds Epo)
Transmembrane alpha-helix
Cytosolic domain (binds JAK kinase)
Each receptor is associated with a JAK kinase (inactive by default)
Epo binding → Receptor dimerization
Dimerization brings JAKs close → Autophosphorylation
JAKs phosphorylate each other on activation lip
Activates kinase activity
Activated JAKs → Phosphorylate tyrosine residues on receptor’s intracellular domain
JAK = tyrosine kinase (targets tyrosine residues only)
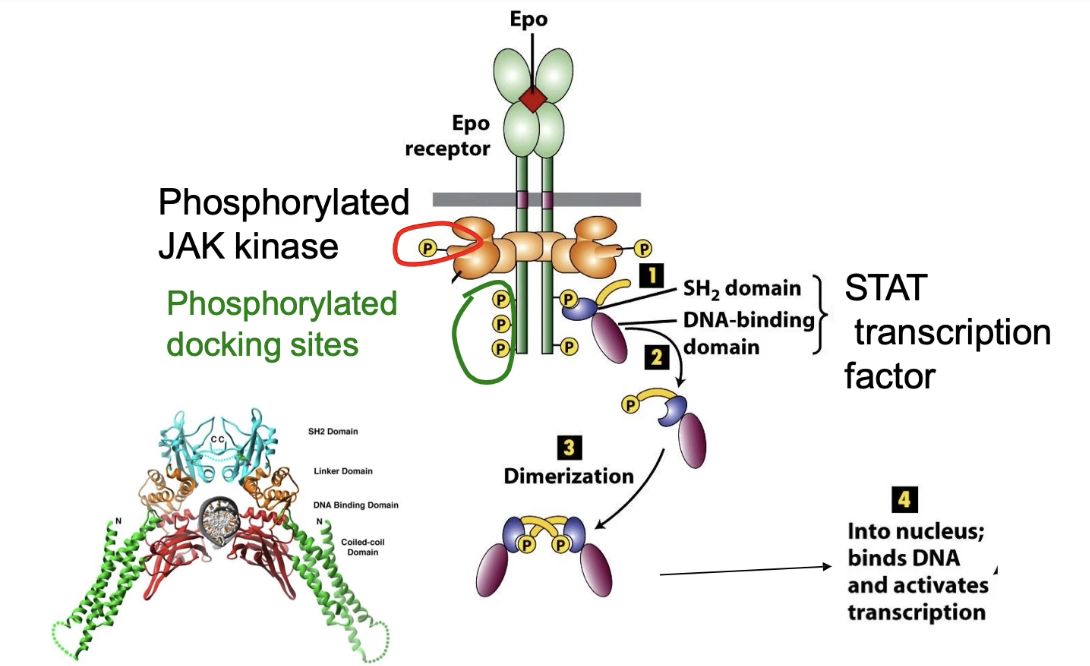
How does activation of the erythropoietin receptor lead to STAT activation and gene expression?
Phosphorylated tyrosines on EpoR = docking sites for other proteins
STAT transcription factors bind using SH2 domain (recognizes phosphotyrosine)
Binding brings STAT near JAK kinase
JAK phosphorylates STAT → activates it
Phosphorylated STAT dimerizes (activation step)
Dimerization exposes nuclear localization sequence
STAT dimer enters nucleus → activates transcription of target genes
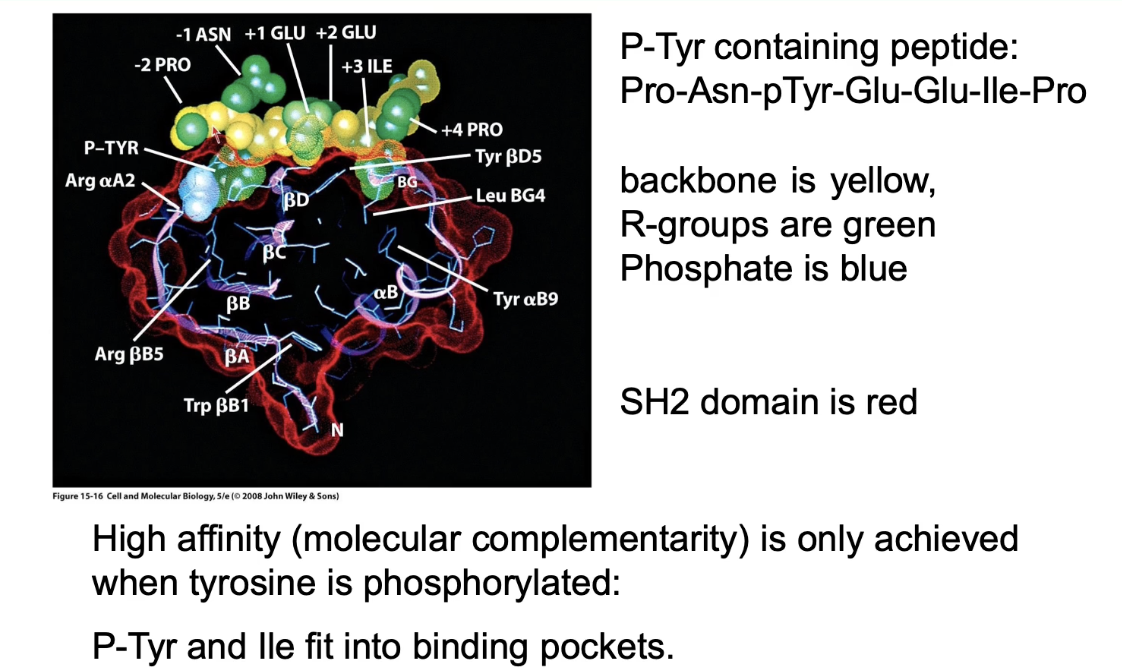
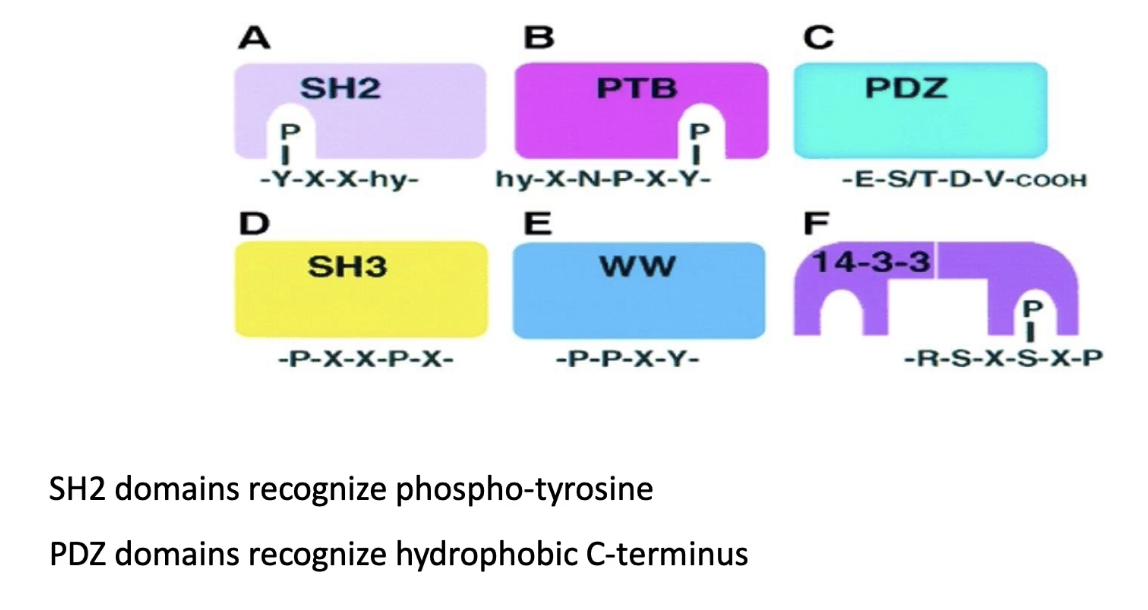
What is the function of the SH2 domain in cytokine signaling?
Protein-protein interaction domain (no enzymatic function)
Binds to specific phosphotyrosine-containing sequences
Binding is high affinity when tyrosine is phosphorylated
Used for:
Re-localizing proteins (e.g., STAT to receptor)
Linking proteins in a signaling pathway
Example binding sequence: Pro-Asn-pTyr-Glu-Glu-Ile-Pro
Binding is reversible depending on phosphorylation state
What are different types of protein-protein interaction domains, and how do they differ?
All link proteins together
Some require reversible modifications (like phosphorylation):
SH2, PTB, 14-3-3: bind phosphorylated tyrosine
Allow reversible binding
Others bind unmodified sequences:
PDZ: binds C-terminal hydrophobic residues
SH3 & WW: bind proline-rich sequences
Binding is not reversible
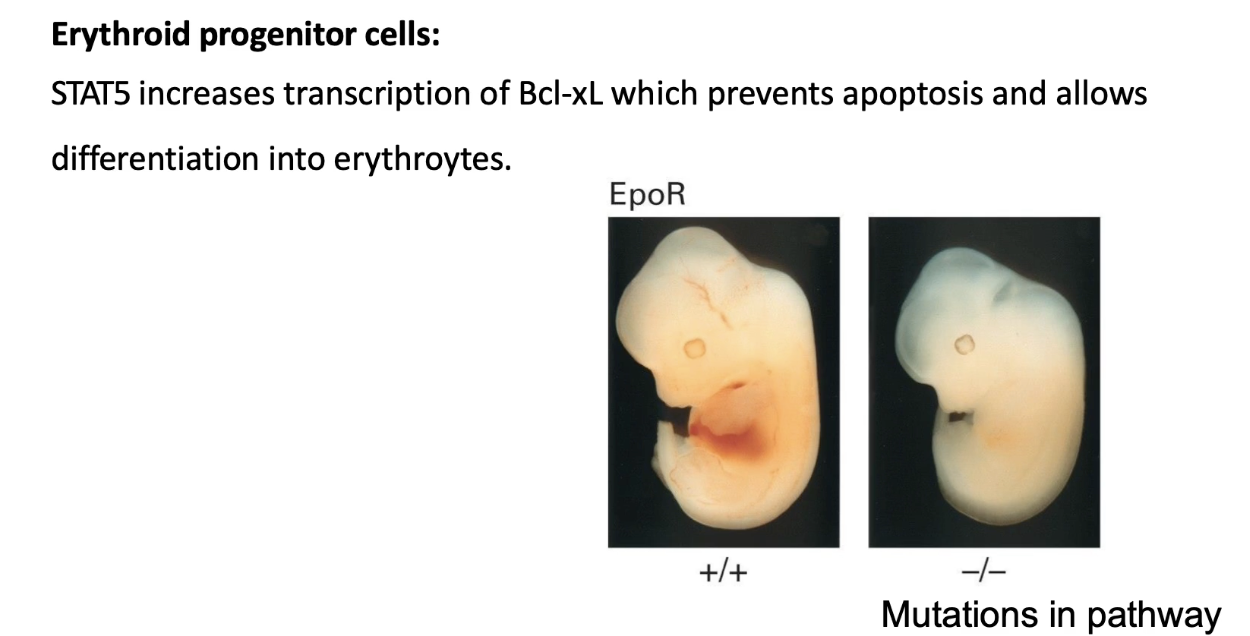
How does the cytokine/JAK-STAT pathway regulate red blood cell production (erythrogenesis)?
Signal: Erythropoietin (Epo)
Receptor: Epo receptor (activates JAK-STAT pathway)
STAT5: Key transcription factor activated
Target gene: Bcl-xL (codes for Bcl-XL protein)
Bcl-XL inhibits apoptosis
Allows erythroid progenitor cells to survive & differentiate
Erythrogenesis sites:
Bone marrow (primary source)
Fetal liver (during development)
Mutation impacts: Defects in Epo, EpoR, JAK, STAT5, or Bcl-xL → no red blood cells produced
Example assay:
WT embryo: red fetal liver (active erythrogenesis)
EpoR knockout: pale liver (no RBC production)
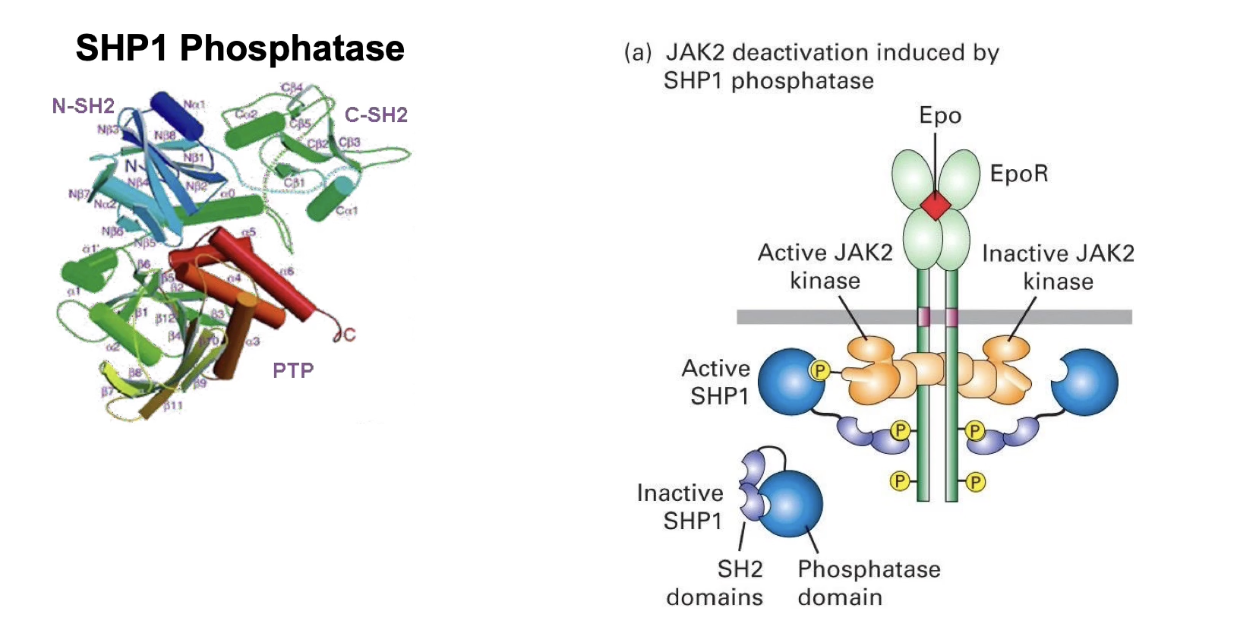
Why and how is the cytokine/JAK-STAT pathway turned off, and what are the risks of failure to regulate it?
Why regulation is critical:
Too little signaling → no red blood cell production → lethal
Too much signaling → overproduction of RBCs → elevated hematocrit
Increases blood viscosity
Risk of capillary blockages, stroke, heart attack
Example:
Athletes may use Epo doping (external Epo) to boost oxygen capacity
Increases endurance but is dangerous & potentially lethal
How the pathway is turned off:
Short-term inactivation via dephosphorylation
Protein: SHP1 phosphatase
Has two SH2 domains → binds to same sites as STAT
Once bound, SHP1 dephosphorylates JAK kinase → turns off signaling
Can be quickly reversed when SHP1 detaches
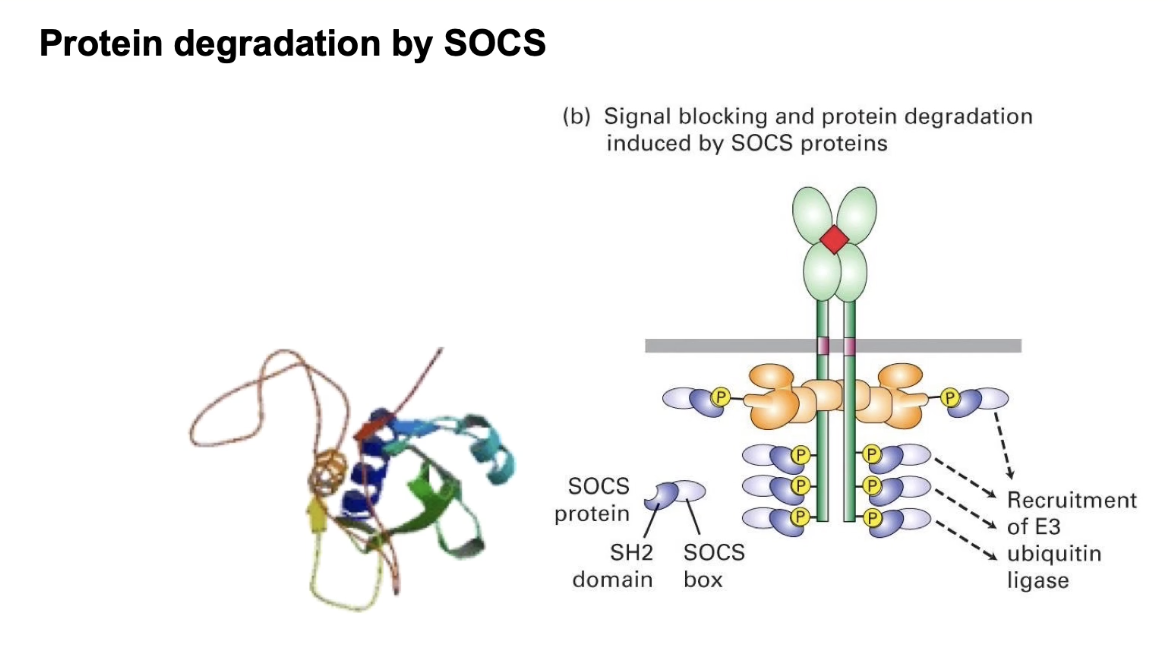
What are the long-term mechanisms that turn off erythrogenesis via the JAK-STAT pathway, and what happens when this regulation fails?
SOCS Protein (Suppressor of Cytokine Signaling):
Binds phosphorylated docking sites via SH2 domain
Expressed in response to high O2 levels
Blocks STAT binding
Acts as an E3 ubiquitin ligase:
Targets JAK kinase for ubiquitination
Leads to proteasomal degradation of JAK
Long-term shutdown (JAK must be re-synthesized to reactivate)
Gene Expression Regulation:
SOCS is upregulated when oxygen levels are high
Acts as a negative feedback loop to stop red blood cell production
Mutations in EpoR gene:
Truncated receptors → shorter docking sites
Decreased sensitivity to SHP1 and SOCS
Results in elevated hematocrit (mimics Epo doping)
Seen naturally in some individuals (not doping)
Receptor recycling:
Endocytosis removes receptor from surface
Ligand (Epo) dissociates → signal ends
Receptor may be recycled but won’t be reactivated unless Epo is present
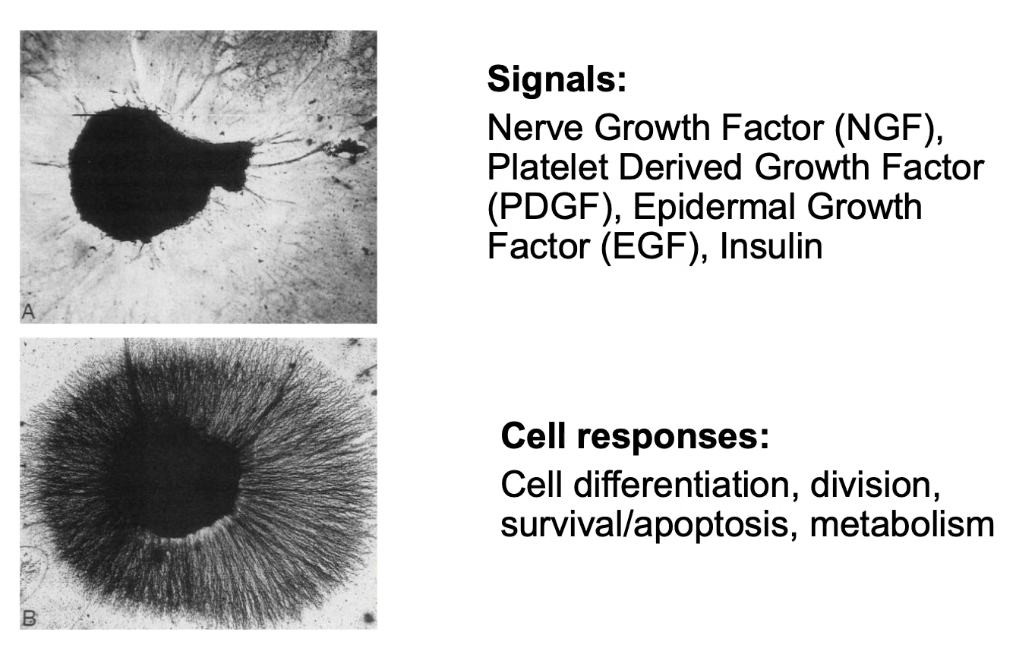
What are Receptor Tyrosine Kinases (RTKs) and what do they do?
RTKs = cell surface receptors with intrinsic tyrosine kinase activity
Activated by hormones: NGF, PDGF, EGF, insulin
Functions: cell differentiation, survival, proliferation, metabolism
Mechanism:
Hormone binds → receptor dimerizes
Activates kinase → phosphorylates tyrosines
Triggers downstream signaling (e.g., Ras G-protein activation)
Example: NGF → neuron axon growth
How do Receptor Tyrosine Kinases (RTKs) activate cell signaling pathways?
RTKs have:
Extracellular signal-binding domain
Single transmembrane domain
Intrinsic kinase activity on cytoplasmic side
Ligand binding → dimerization → autophosphorylation
Activates downstream signaling via Ras G-protein (intracellular, membrane-anchored protein)
Key players that regulate and link Ras to the activated RTK:
GRB2 (adaptor protein)
Ras effectors
GEF (activates Ras)
GAP (inactivates Ras)
Activated Ras → MAP kinase cascade
MAP kinase → phosphorylates transcription factors → changes gene expression
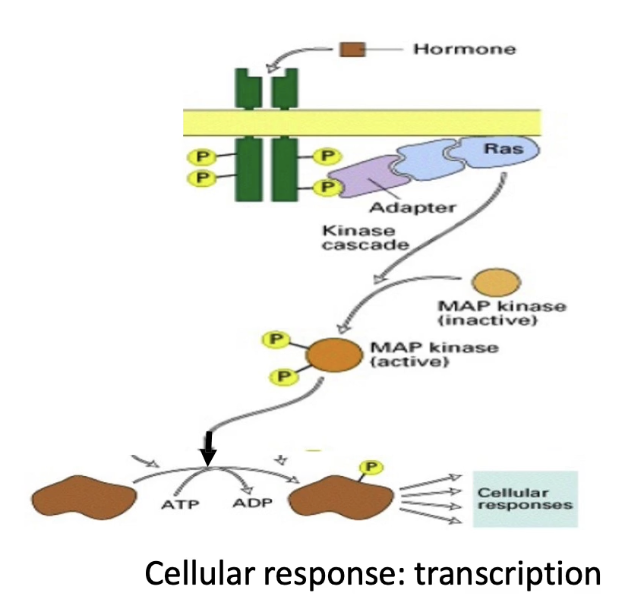
How is RTK activation similar to cytokine receptor activation?
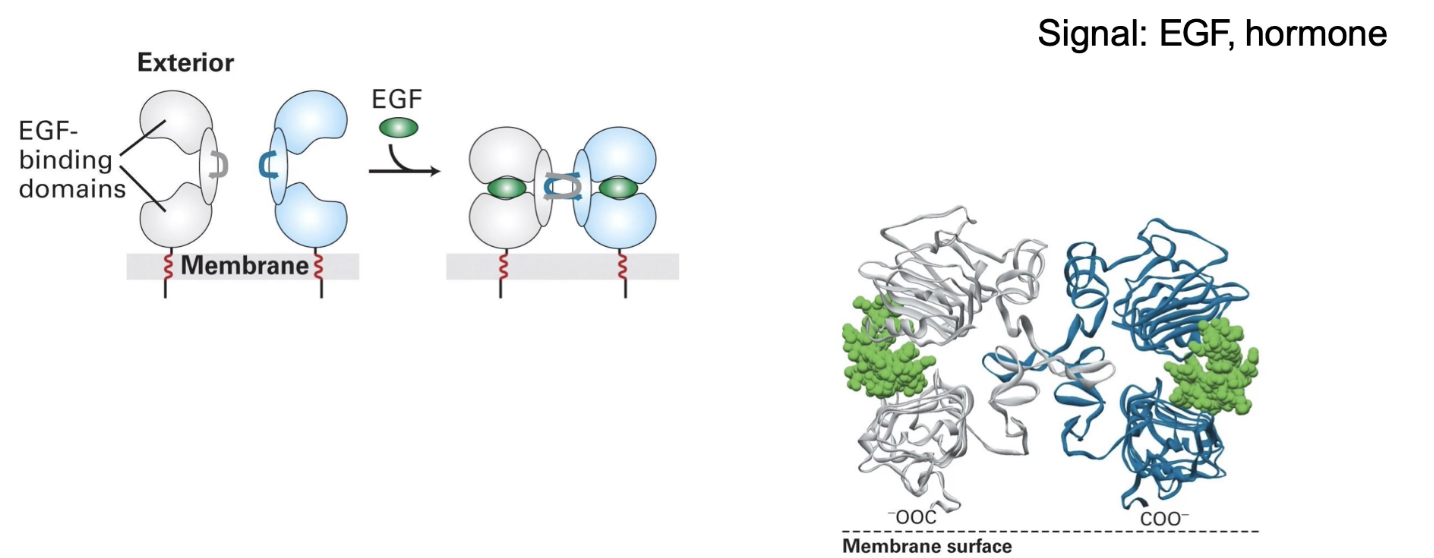
Ligand binding (e.g., EGF) initiates activation
Causes conformational change in extracellular domain
Leads to dimerization of two RTK molecules
Dimerization is necessary for activation of kinase activity
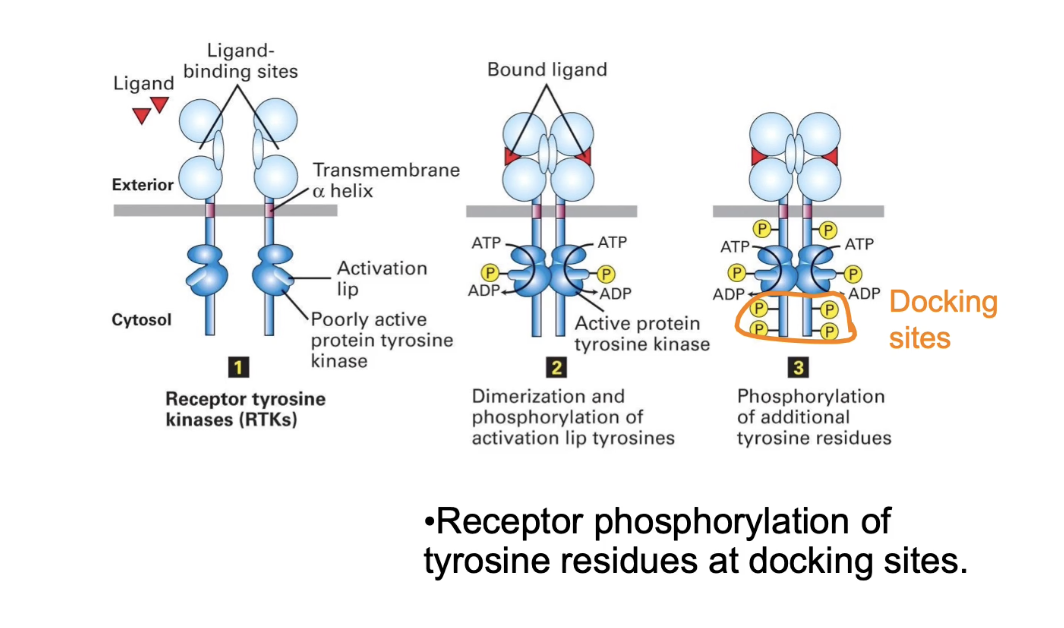
How does dimerization activate Receptor Tyrosine Kinases (RTKs)?
RTK monomers have low intrinsic kinase activity
Dimerization brings kinase domains close together
Enables autophosphorylation of each other's activation lip
Phosphorylation of activation lip increases kinase activity
Activated RTKs phosphorylate tyrosine residues on intracellular domains
These phosphorylated sites serve as docking sites for proteins with SH2 or PTB domains
What is the role of adaptor proteins in RTK signaling pathways?
Adaptor proteins have multiple protein interaction domains
Link activated RTK receptors to downstream signaling proteins
Bind phosphorylated docking sites on RTKs and other signaling proteins
Help bridge signaling components without enzymatic activity
Scaffold proteins are larger adaptor proteins that organize multiple signaling proteins into a structured sequence
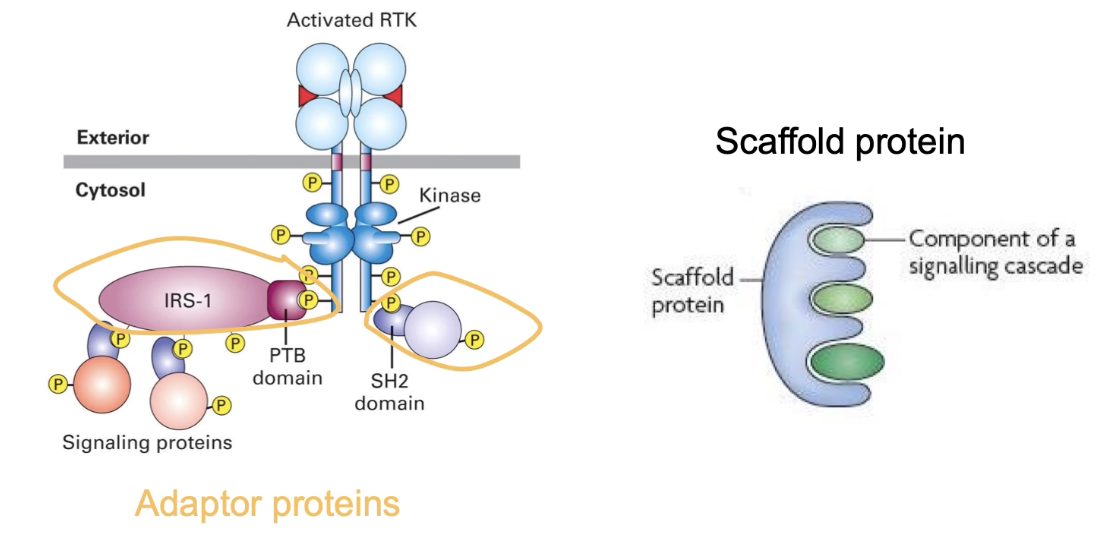
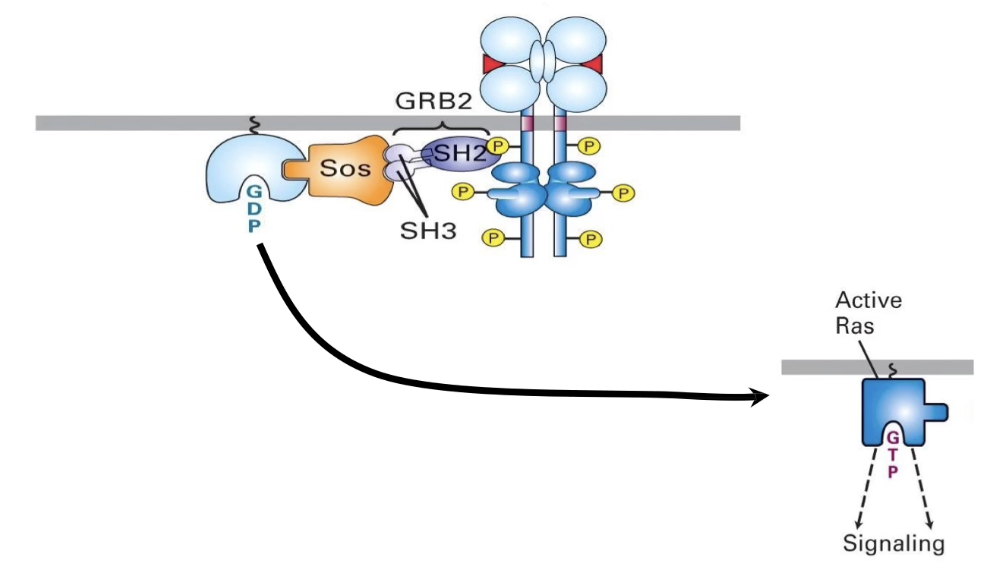
What is GRB2 and how does it function in RTK signaling?
GRB2 is an adaptor protein in RTK signaling
Has 3 domains:
1 SH2 domain → binds phosphorylated tyrosine on RTK docking site (reversible)
2 SH3 domains → bind proline-rich sequences (like in SOS)
Links RTK receptor to SOS, a guanine nucleotide exchange factor (GEF)
Facilitates activation of Ras by recruiting SOS to the membrane
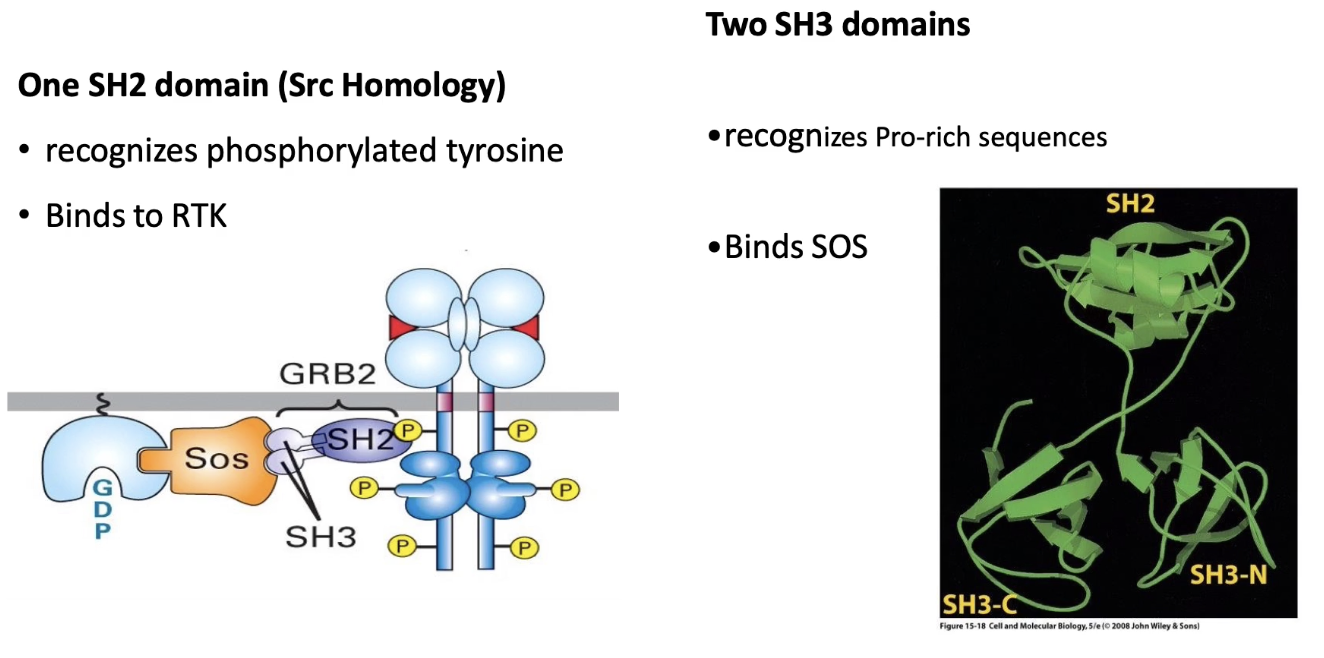
What is the role of the SH3 domain in RTK signaling?
SH3 domain is a protein-protein interaction domain
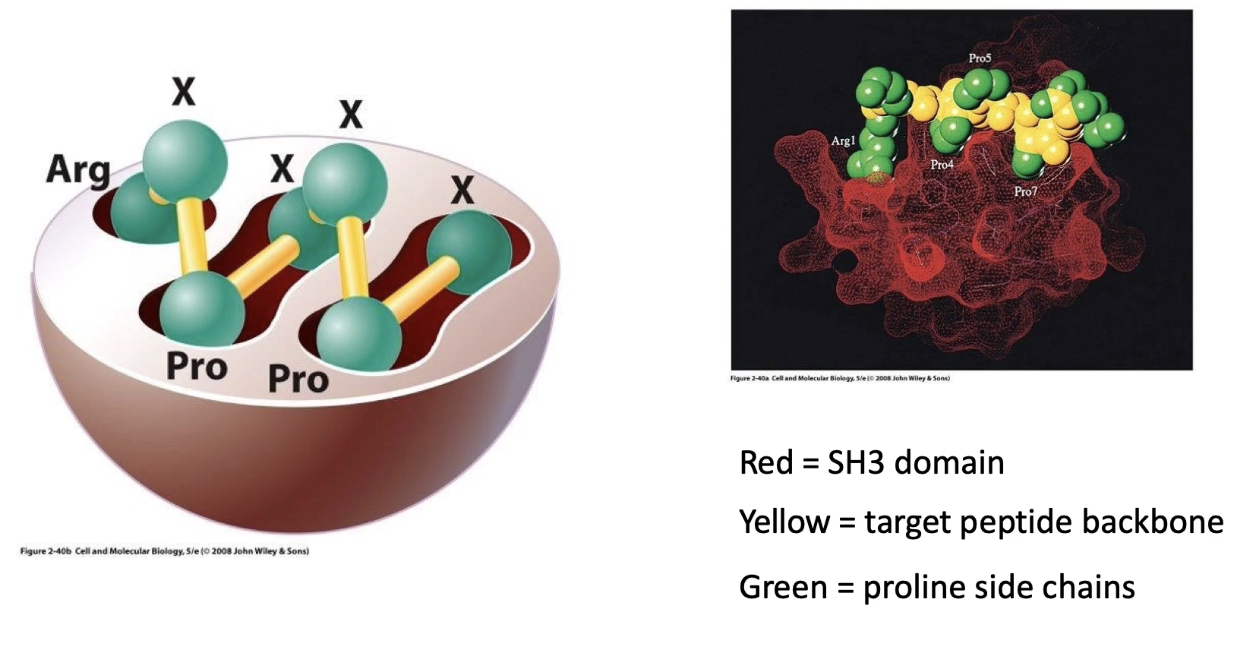
Binds to proline-rich sequences on target proteins (e.g., SOS)
Interaction is based on molecular complementarity:
SH3 domain (red) fits into clefts of proline-rich peptide (green)
Binding is specific and stable, not dependent on phosphorylation
Enables adaptor proteins (like GRB2) to link RTKs to downstream signaling components
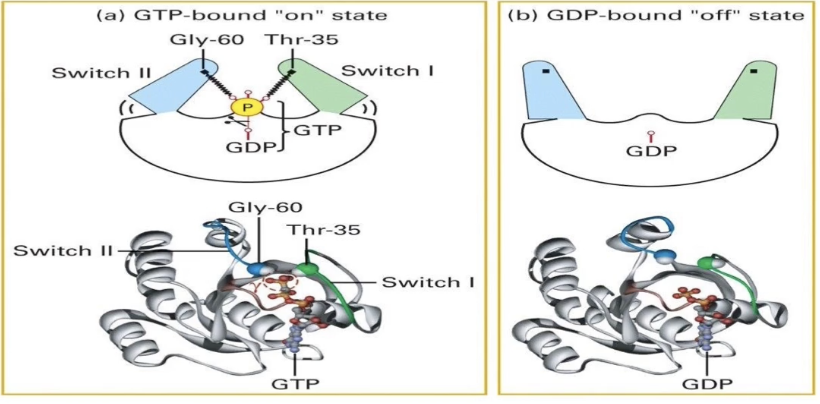
How does the Ras G-protein cycle between active and inactive states?
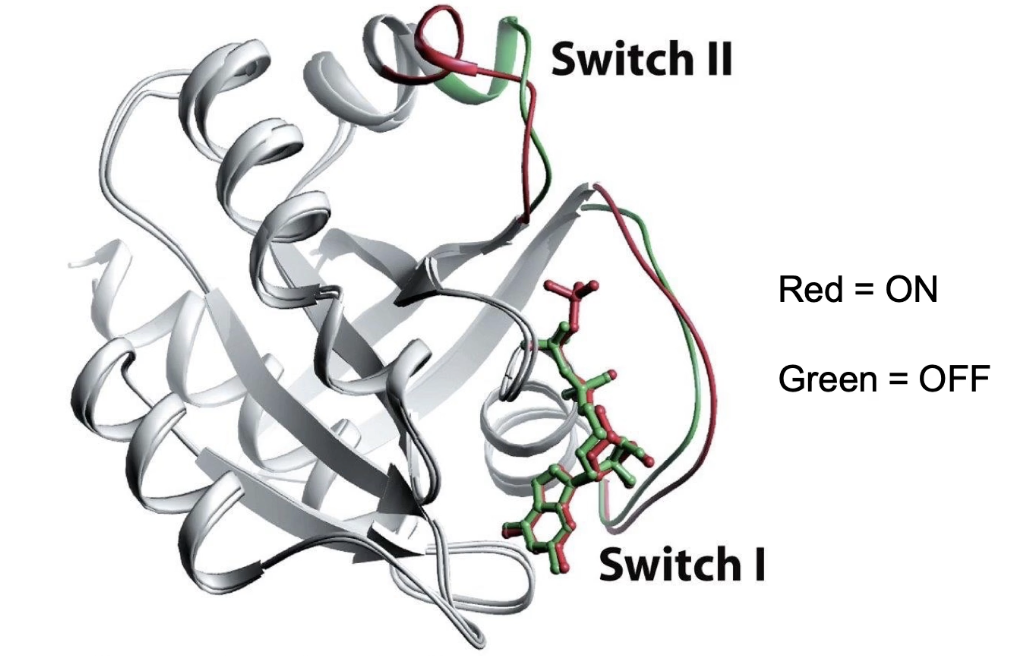
Ras is a GTPase switch protein: active when bound to GTP, inactive when bound to GDP
GTP-bound ("ON" state):
GTP’s terminal phosphate interacts with glycine and threonine residues on the switch regions
Switches are pulled together, stabilizing the active conformation
Intrinsic GTPase activity:
Always active but not regulated ON/OFF
Hydrolyzes GTP to GDP + Pi, turning Ras "OFF"
GDP-bound ("OFF" state):
Loss of terminal phosphate causes switches to fold out
Glycine and threonine residues no longer interact with GDP
GDP has low affinity and dissociates, allowing new GTP to bind and reactivate Ras
This cycle regulates Ras activity in RTK signaling pathways
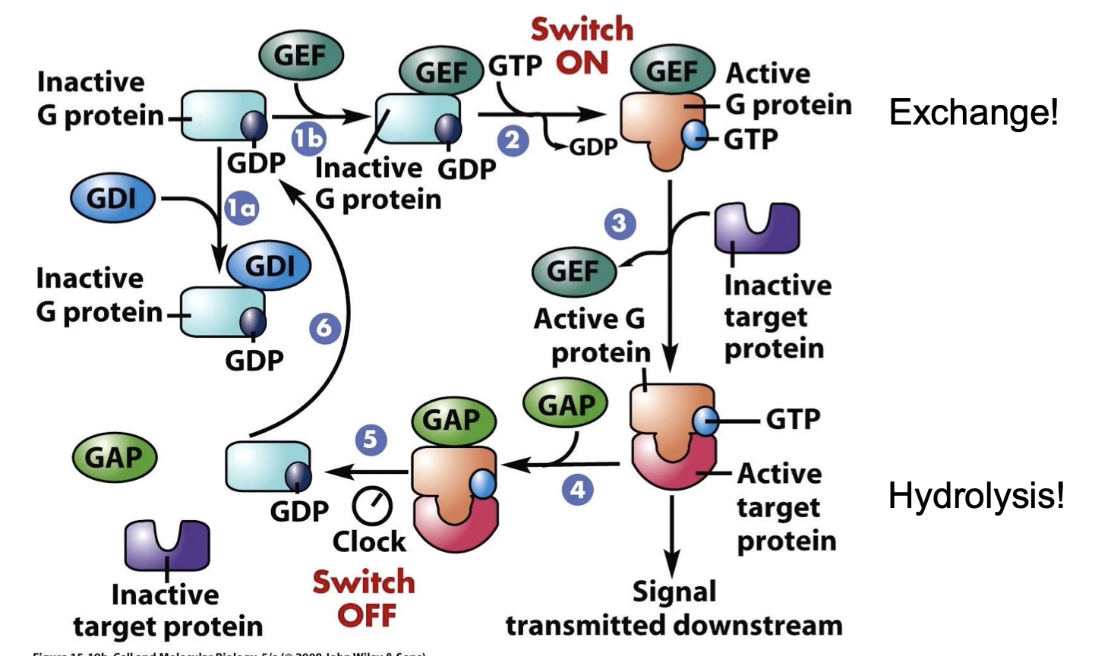
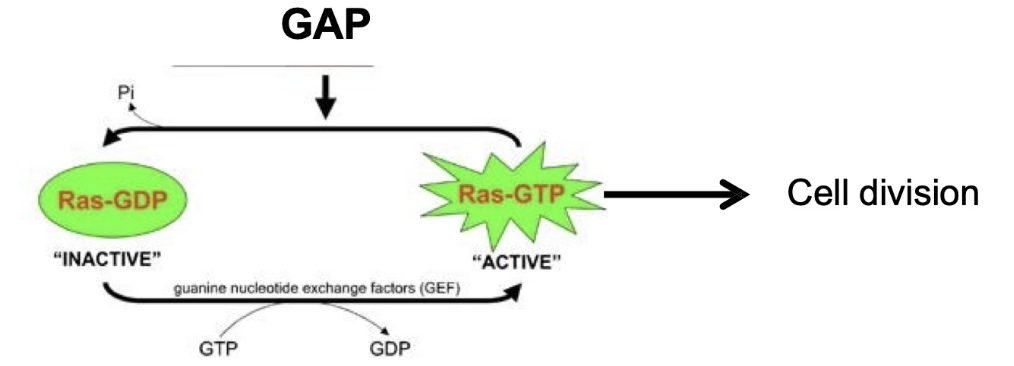
What proteins regulate the activation and inactivation of G-proteins?
GEF (Guanine Nucleotide Exchange Factor):
Promotes GDP release
Facilitates GTP binding → activates G-protein
GAP (GTPase Activating Protein):
Enhances intrinsic GTPase activity (up to 100x)
Accelerates GTP hydrolysis → inactivates G-protein
GDI (Guanine Nucleotide Dissociation Inhibitor):
Increases GDP affinity to nucleotide-binding pocket
Keeps G-protein in inactive “OFF” state
How is Ras G-protein activity regulated in the RTK pathway?
OFF = GDP-bound (inactive), stabilized by GDI
GEF triggers GDP release → GTP binds → ON (active) state → G-protein interacts with target protein
GAP accelerates GTP hydrolysis → returns to OFF state → can’t interact with target protein
Duration of ON state controlled by GAP → affects signaling length
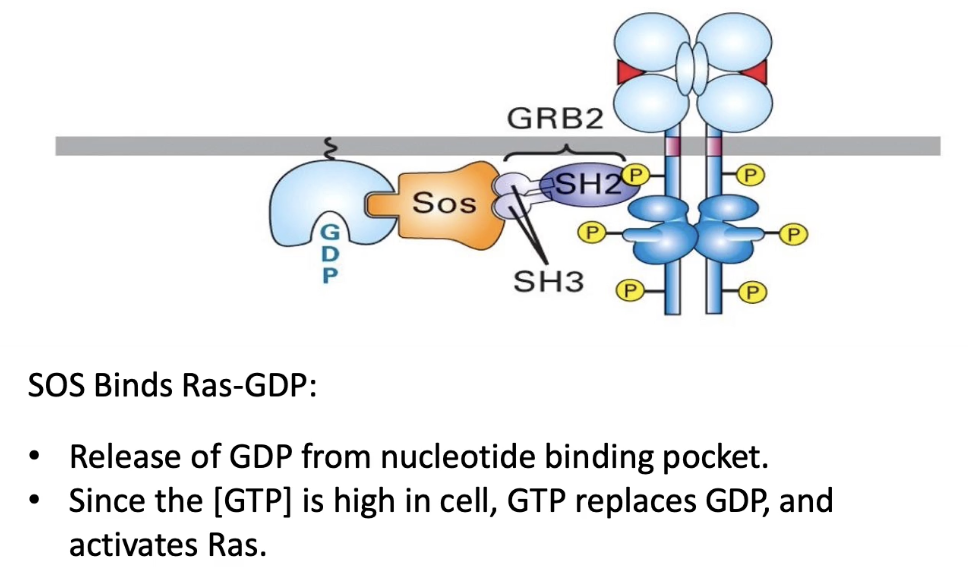
How is Ras G-protein activated by SOS in the RTK pathway?
Activated RTK recruits adaptor protein GRB2
GRB2’s SH3 domains bind SOS, bringing it to the membrane
SOS (a GEF) interacts with membrane-bound Ras
SOS promotes GDP release from Ras → GTP binds
Ras becomes activated (ON state)
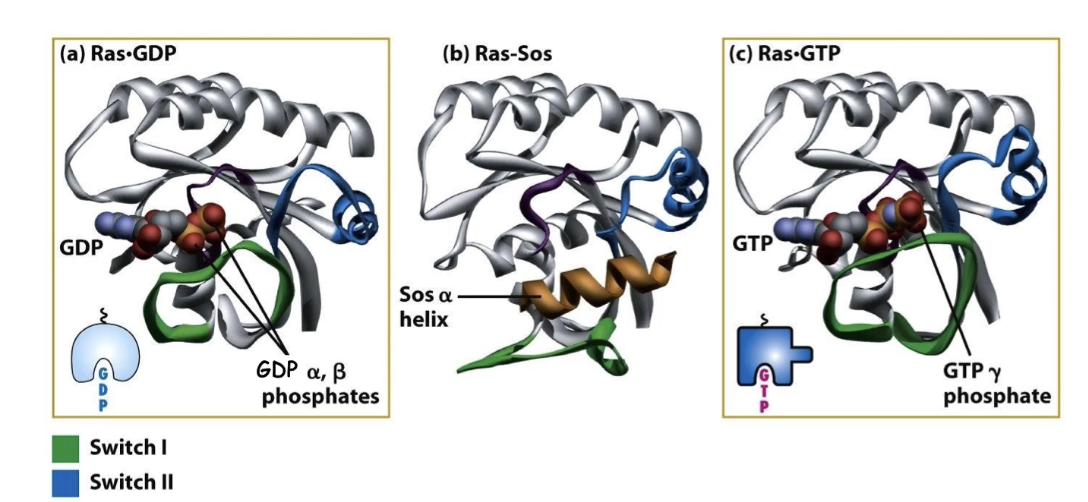
What are the three conformational states of Ras protein?
Inactive: Ras bound to GDP
Intermediate: SOS binds Ras, displacing GDP
Active: Ras bound to GTP
What is the role of NF1 in regulating Ras activity, and what happens when NF1 is mutated?
NF1 is a GAP protein that enhances Ras’s intrinsic GTPase activity, speeding up GTP hydrolysis and inactivating Ras.
Presence of NF1 shortens Ras active time, turning signaling off faster.
Loss of NF1 function removes GAP activity, causing Ras to stay active longer.
Prolonged Ras activity leads to excessive signaling, increasing cell division rates.
NF1 mutations are linked to tumor formation in neural tissue (neurofibromatosis 1).
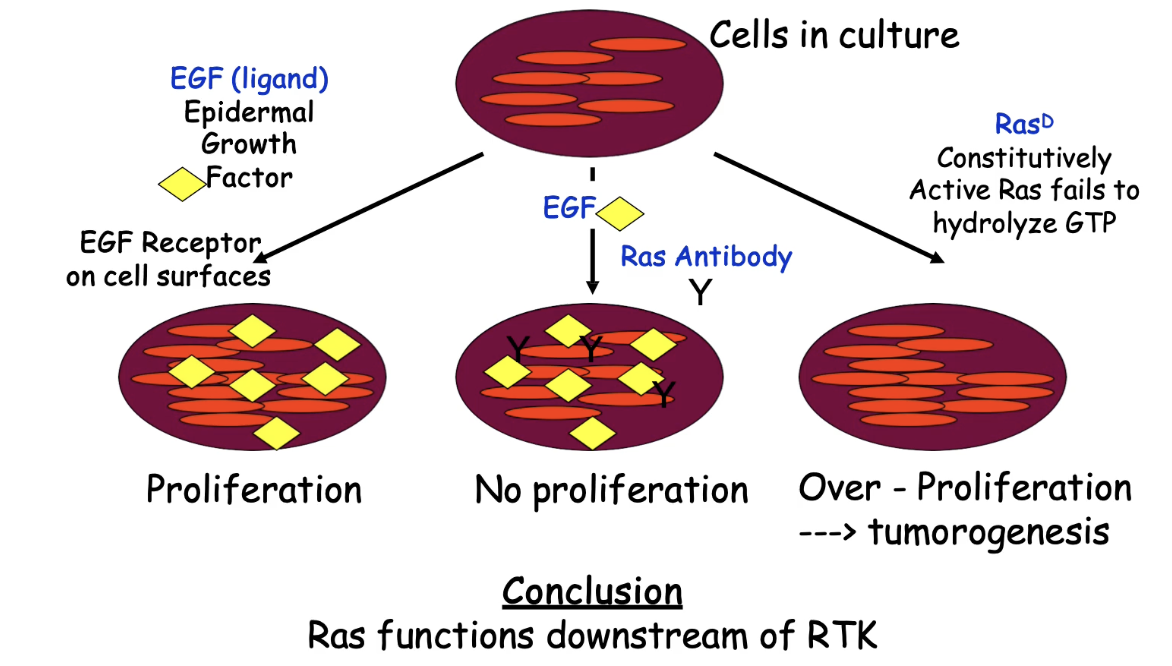
How does Ras activation relate to RTK signaling and cell division?
RTK activation is upstream of Ras activation
Ras activation occurs after RTK is activated by ligand (e.g., EGF)
Ras links RTK signaling to changes in gene expression via downstream pathways
Key experiment findings:
EGF only → RTK activated → cell division (control)
Anti-Ras antibody + EGF → Ras blocked → no cell division
Shows Ras is required downstream of RTK
Ras-D (always active) + no EGF → cell division
Shows Ras can bypass RTK if always active
Conclusion:
Ras is downstream of RTK
Ras activation alone can trigger cell division
Constitutively active Ras (Ras-D) mimics tumor-like overproliferation
Dominant Ras mutations are linked to cancer in various cell types
How do mutations in the RTK-Ras pathway lead to cancer?
Constitutively active Ras → continuous signaling → uncontrolled cell division
Example: loss of glycine in Ras blocks GAP binding, so Ras stays ON
Her2 mutant (RTK) → always dimerized → active without EGF
Leads to persistent signaling → hereditary breast cancer
NF1 loss-of-function → no GAP activity → Ras not turned OFF → overproliferation
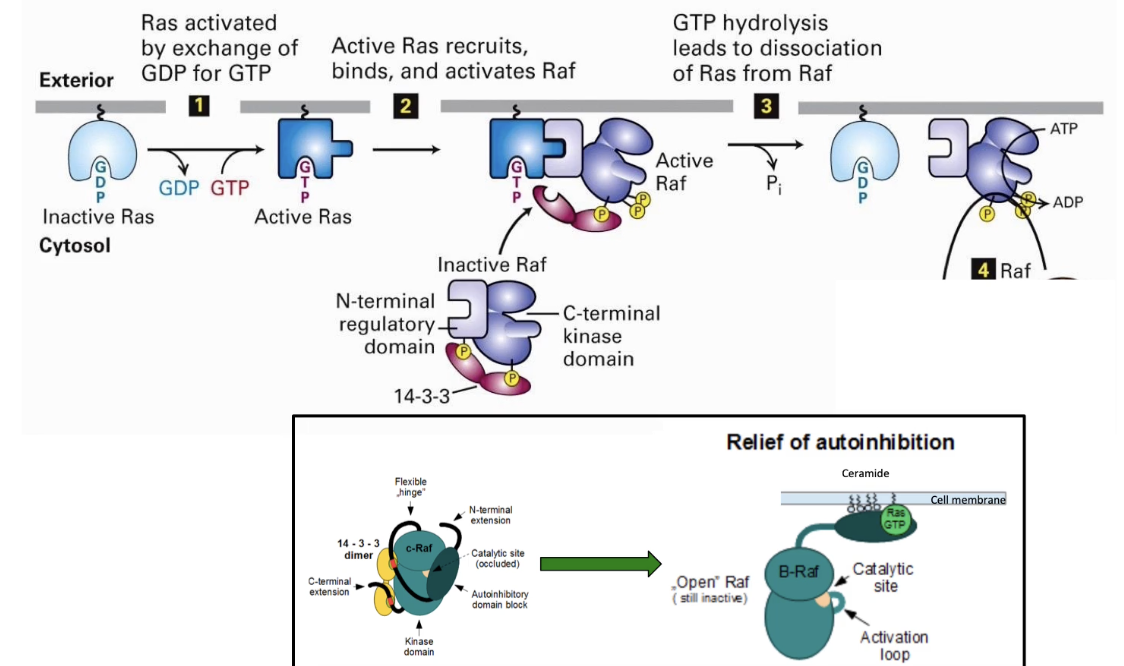
What happens after Ras is activated in the RTK signaling pathway?
Activated Ras binds and activates Raf (a serine/threonine kinase)
Raf is normally inhibited by binding to 14-3-3 adaptor protein
Ras binding causes release of 14-3-3 → Raf activation
Raf activation begins the MAP kinase cascade, leading to changes in gene expression and cell behavior
What is the kinase cascade activated by Ras in the RTK pathway?
Ras activates Raf (MAP kinase kinase kinase)
Raf phosphorylates MEK (MAP kinase kinase)
MEK phosphorylates MAP kinase at 2 residues, tyrosine & threonine
Activated MAP kinase:
Dimerizes
Moves to nucleus
Phosphorylates transcription factors → changes in gene expression
Scaffold proteins help organize and stabilize the kinase cascade
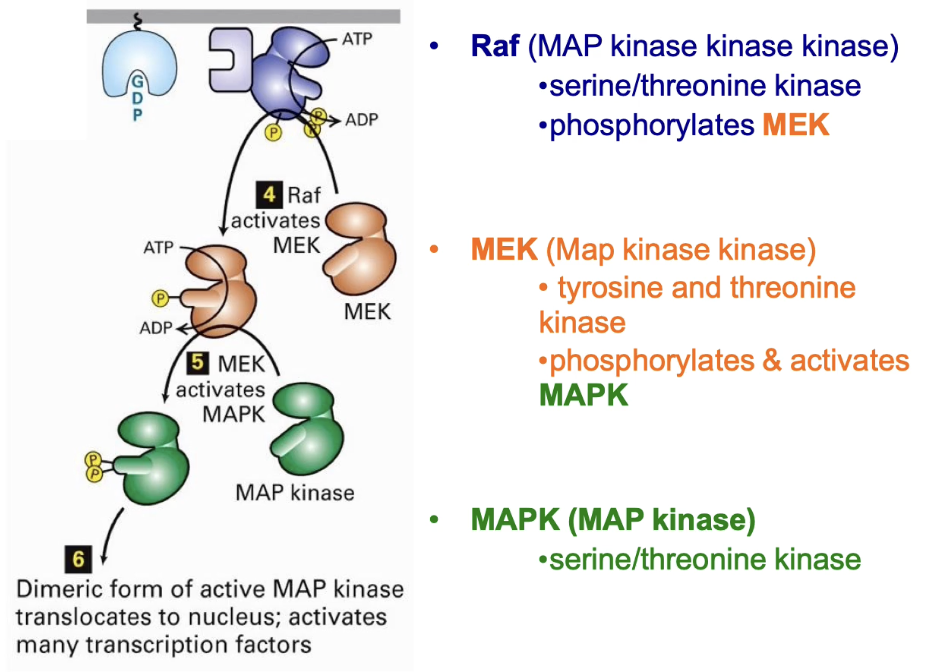
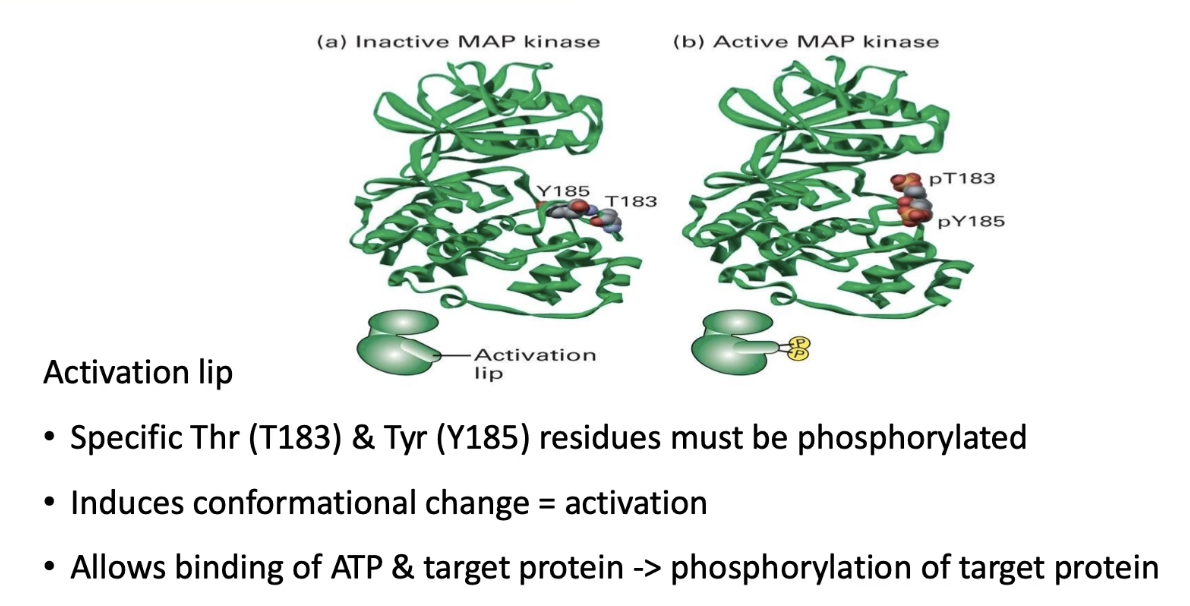
How is MAP kinase activated in Ras-linked RTK signaling pathways?
Activated by MEK, a dual-specificity kinase (targets threonine & tyrosine)
Phosphorylation occurs on the activation lip of MAP kinase
Conformational change exposes ATP & substrate binding pockets
Similar activation mechanism as seen in other kinases
MAP kinase = final downstream target in Ras-linked RTK pathways
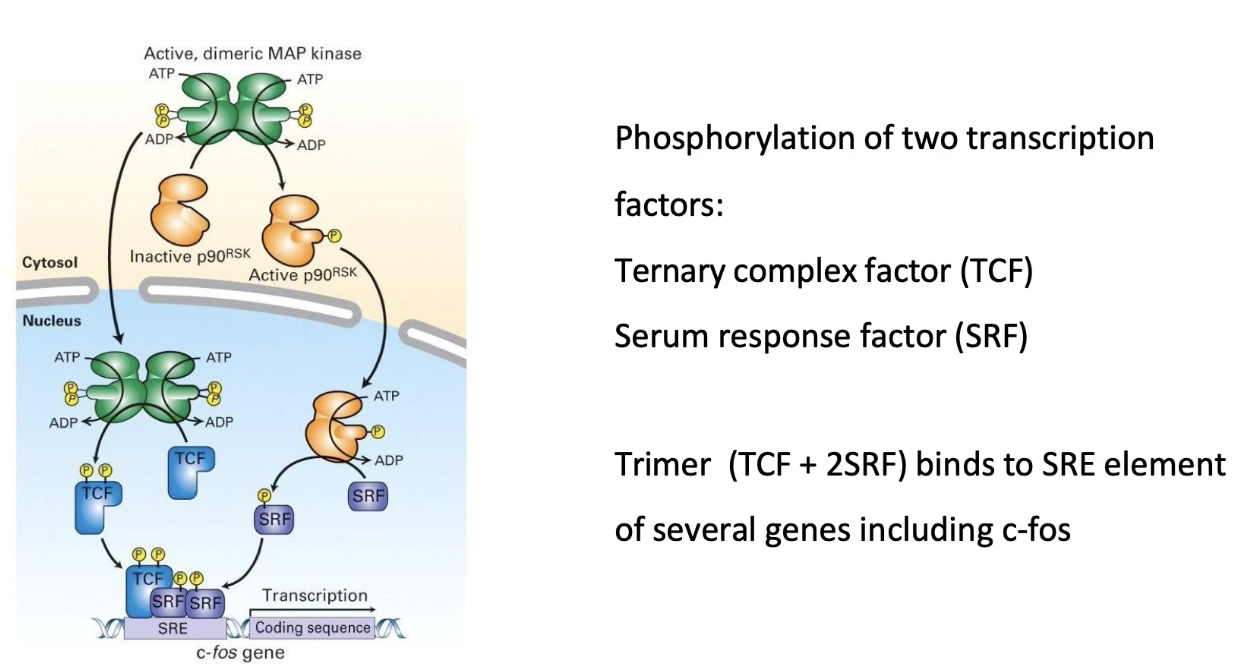
What does activated MAP kinase do in the Ras-RTK pathway?
Phosphorylates P90 RSK in cytoplasm → RSK moves into nucleus
MAP kinase also enters nucleus
In nucleus:
MAP kinase phosphorylates TCF (ternary complex factor)
P90 RSK phosphorylates SRF (serum response factor)
TCF + SRF bind to SRE (Serum Response Element) on DNA
This activates transcription of genes like C-fos
C-fos promotes transcription of cell cycle genes
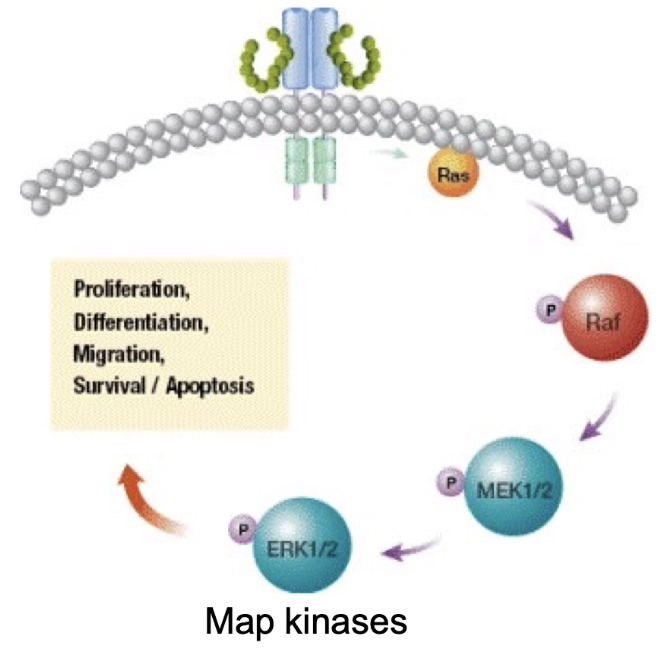
What is the final outcome of RTK signaling and how does signal amplification work?
Final outcome: Activation of gene transcription → cell division, differentiation, other behaviors
Signal amplification:
Each kinase can activate many target proteins
Example: One EGF molecule → millions of active MAP kinases (ERK1/2)
Leads to millions of copies of proteins needed for cell division
Sensitivity: Pathway responds to very low hormone levels (nanomolar or 10-9 M range)
What are GPCRs and why are they important in medicine?
GPCRs = large family of receptors in humans
Involved in almost every physiological process
Many diseases linked to GPCR dysfunction
Most pharmaceuticals target GPCRs to treat diseases
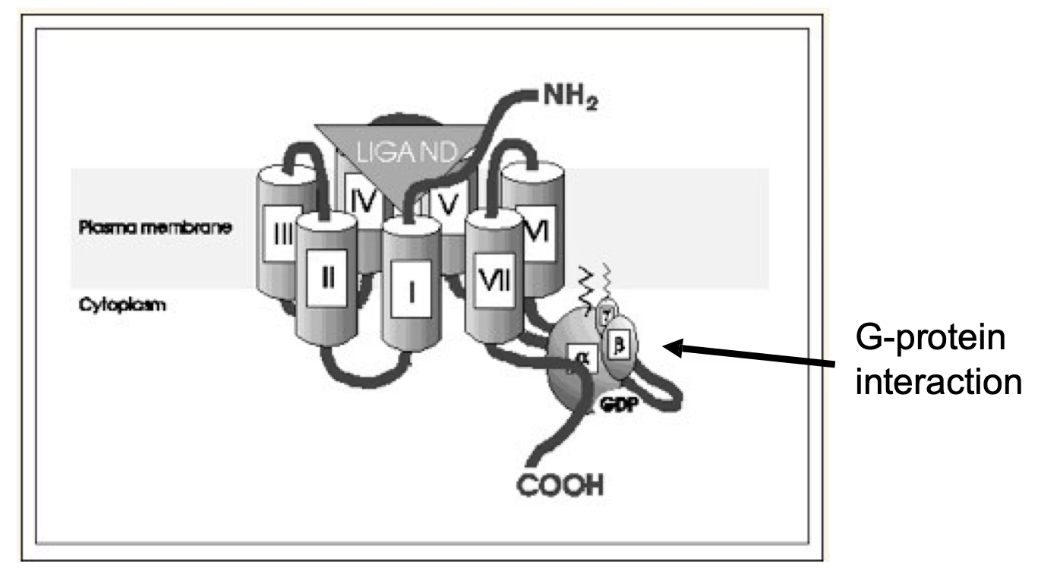
What is the common structure of GPCRs and what do their segments do?
7 transmembrane alpha helices
4 extracellular loops (E1–E4) form the signal-binding domain
4 cytoplasmic loops (C1–C4) interact with trimeric G protein
Examples: stress receptors, rhodopsins (vision), odorant receptors, hormone/neurotransmitter receptors, plant growth hormone receptors, yeast glucose sensor
~800 GPCR genes in humans (~4% of protein-coding genome)
Many diseases linked to GPCR dysfunction
What role do GPCRs play in the mammalian stress response involving catecholamines?
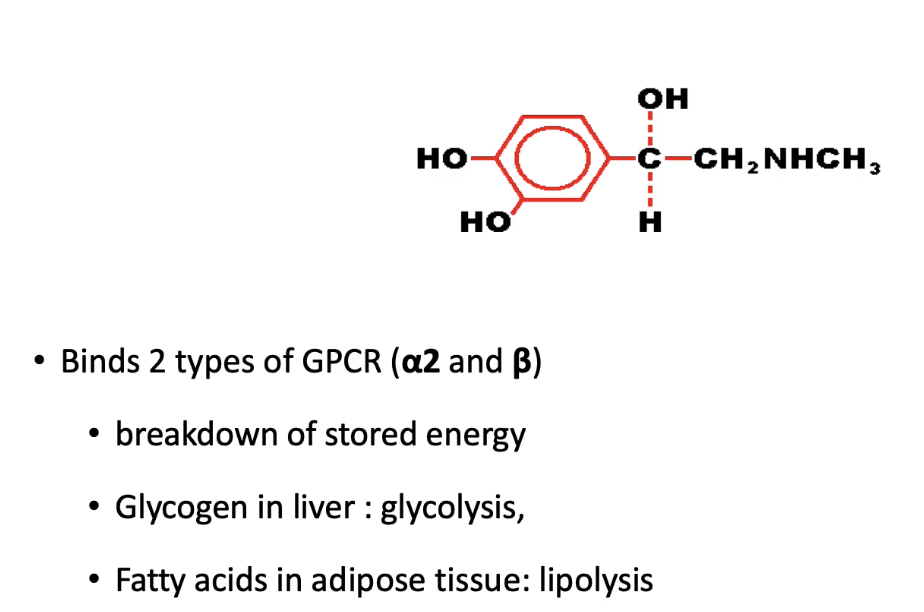
Catecholamines (water-soluble signals that circulate bloodstream): epinephrine, norepinephrine, dopamine (fight-or-flight hormones)
Catecholamine receptors are GPCRs
Activation triggers trimeric G-protein → activates adenylyl cyclase (effector protein)
Adenylyl cyclase increases cAMP (secondary messenger)
Increased cAMP triggers:
Fast response: enzyme activation (quick energy release)
Slow response: gene transcription activation

How do alpha-2 and beta adrenergic receptors respond to epinephrine in different tissues?
Beta adrenergic receptors (stimulatory):
Liver & adipose: stimulate glycolysis & lipolysis (fuel mobilization)
Heart muscle: increase contraction (more blood supply)
Intestinal smooth muscle: cause relaxation (reduce energy use in digestion so it can be focused on on locomotory muscles)
Alpha-2 adrenergic receptors (inhibitory):
Found in blood vessels in skin, kidneys, smooth muscles of intestines
Cause artery constriction and reduce blood flow to periphery
Overall: Coordinated responses increase energy supply to stress-response tissues.
Where do catecholamines like epinephrine and norepinephrine come from, and what roles do they play in the stress response?
Catecholamines come from:
Adrenal glands (small endocrine glands that sit on top of kidneys) produce epinephrine (adrenaline) and steroids like aldosterone & cortisol.
Norepinephrine is secreted by nerve cells and acts as a neurotransmitter.
Function of epinephrine:
Binds both beta and alpha-2 adrenergic receptors, causing different responses depending on receptor type.
Triggers stress response by increasing ATP supply through breakdown of energy stores, glycogen and triacylglycerol:
Glycolysis in liver (breaking down glycogen to glucose)
Lipolysis in adipose tissue (breaking down fatty acids).
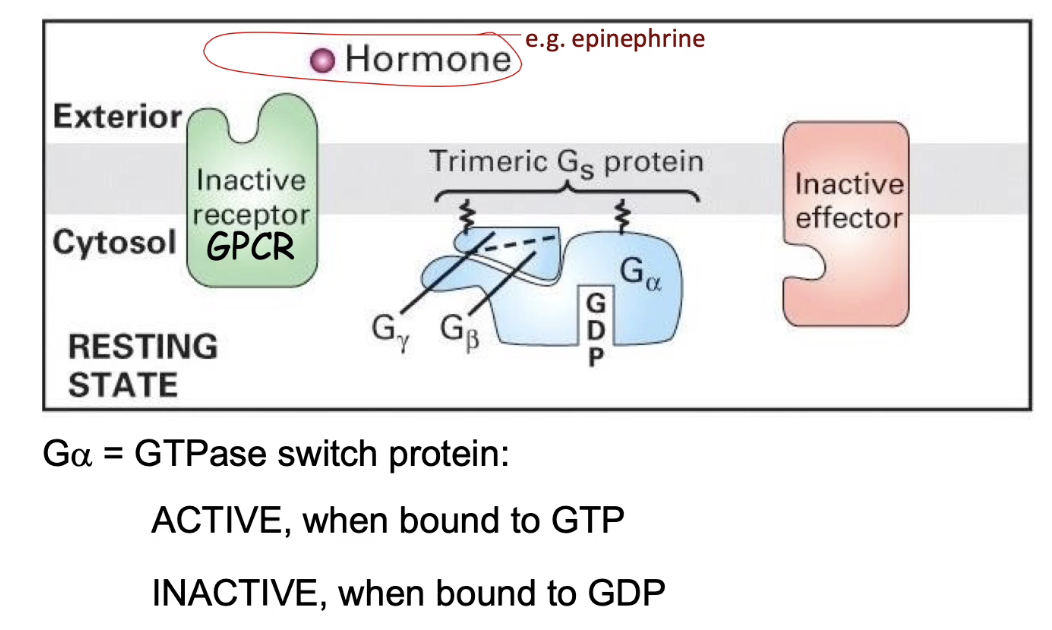
How is a GPCR signaling pathway activated, and what role does the trimeric G-protein play?
Inactive GPCR: Not bound to signal; not associated with trimeric G-protein.
Trimeric G-protein: Lipid-anchored, with 3 subunits (α, β, γ);
Inactive when bound to GDP
Active when bound to GTP
Activation:
Signal binds to GPCR → GPCR undergoes conformational change
GPCR activates the G-protein by promoting GDP → GTP exchange
Activated G-protein then interacts with effector protein to trigger downstream effects.
How is a GPCR activated and how does it activate the trimeric G-protein?
Signal binds GPCR → conformational change in intracellular domain
GPCR binds trimeric G-protein with high affinity
This causes GDP to dissociate from the G-protein
GTP binds in place of GDP → G-protein becomes active
`Activated G-protein can now initiate downstream signaling
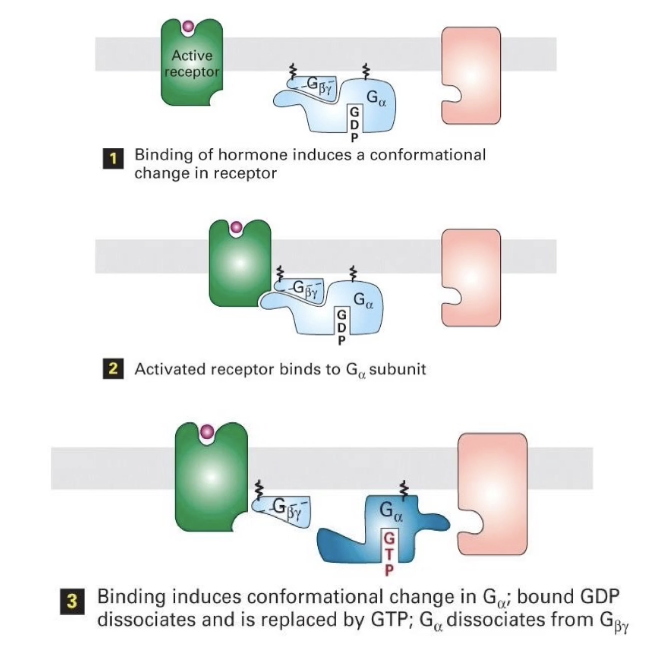
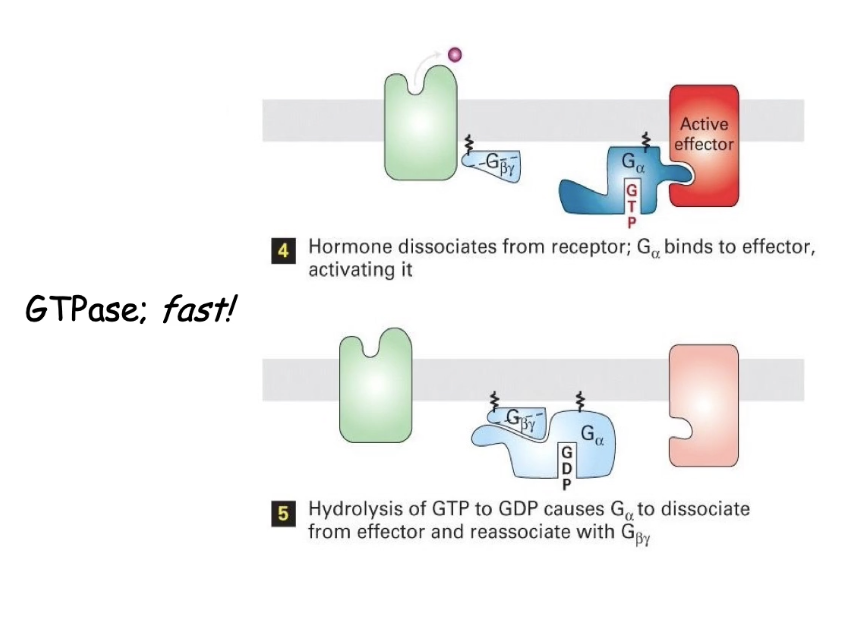
What happens after GPCR activates the trimeric G-protein, and how is the signal turned off?
GTP-bound G-alpha subunit dissociates from beta & gamma
G-alpha activates effector enzyme (e.g., adenylyl cyclase)
Effector stays active while G-alpha is bound with GTP
Intrinsic GTPase of G-alpha hydrolyzes GTP → GDP
G-alpha becomes inactive, releases effector
G-alpha rejoins beta/gamma subunits → signal is off
How do beta-adrenergic receptors activate cAMP production through Gs protein?
Beta-adrenergic receptors activate Gs protein (stimulatory G protein)
Gs alpha subunit (GTP-bound) activates adenylyl cyclase
Adenylyl cyclase increases cAMP (secondary messenger)
GTP → GDP hydrolysis inactivates Gs alpha → stops adenylyl cyclase
cAMP levels drop without continuous receptor activation
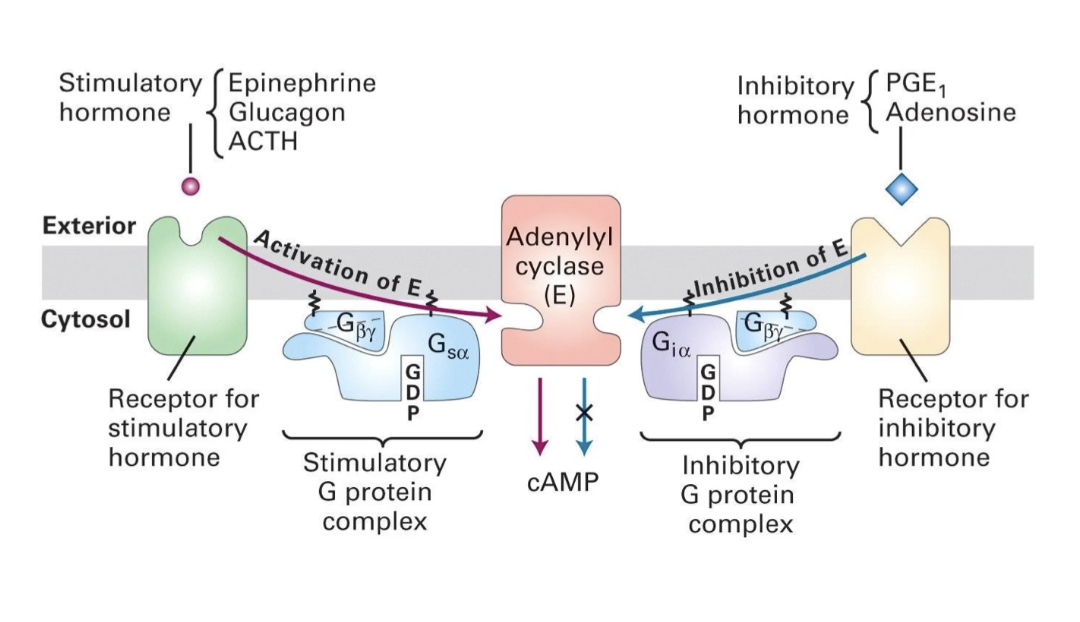
How do alpha2-adrenergic receptors inhibit cAMP production via Gi protein?
Alpha2-adrenergic receptors activate Gi protein
Gi has the same beta & gamma subunits as Gs, but a different alpha subunit (Gi alpha)
Gi alpha (GTP-bound) interacts with different region and inhibits adenylyl cyclase catalytic domain
This prevents cAMP production
Result: No increase in cellular energy signal (cAMP stays low)
How do β-adrenergic and α2-adrenergic receptors differently regulate cAMP levels?
β-adrenergic receptor → activates Gsα → stimulates adenylyl cyclase → ↑ cAMP
α2-adrenergic receptor → activates Giα → inhibits adenylyl cyclase → ↓ cAMP
Epinephrine can bind both receptors to create diverse, tissue-specific responses
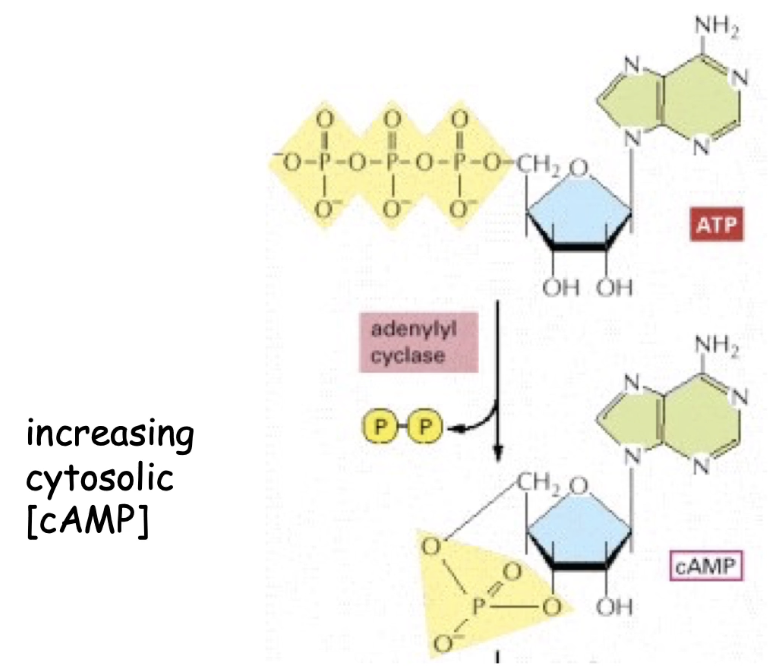
How does adenylyl cyclase produce cyclic AMP (cAMP)?
Converts ATP → cAMP
Releases diphosphate (PPi) as a byproduct
Active adenylyl cyclase = high intracellular cAMP levels
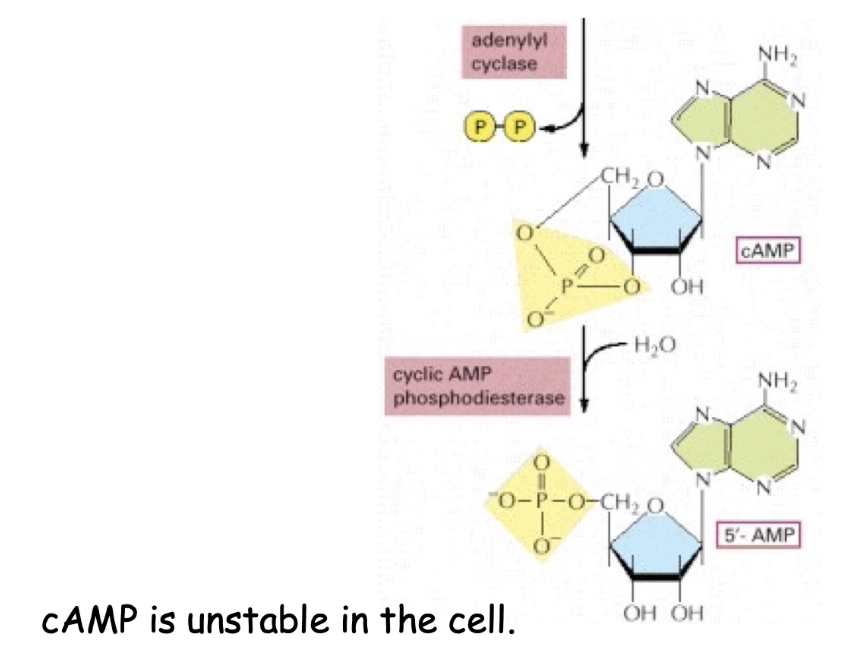
What role does phosphodiesterase play in cAMP regulation?
Breaks down cAMP → 5’AMP
Constitutively active, making cAMP unstable
If adenylyl cyclase is inhibited, cAMP levels drop quickly due to phosphodiesterase
What does cyclic AMP (cAMP) do in GPCR signaling?
Acts as a secondary messenger
Concentration reflects GPCR activity
Modulates target protein activity, especially Protein Kinase A (PKA)
PKA = serine/threonine kinase → phosphorylates many target proteins
How is Protein Kinase A (PKA) activated by cAMP?
Inactive PKA = tetramer (2 regulatory + 2 catalytic subunits)
Low cAMP: regulatory subunits block catalytic activity
High cAMP: cAMP binds regulatory subunits → conformational change
Catalytic subunits released → active PKA
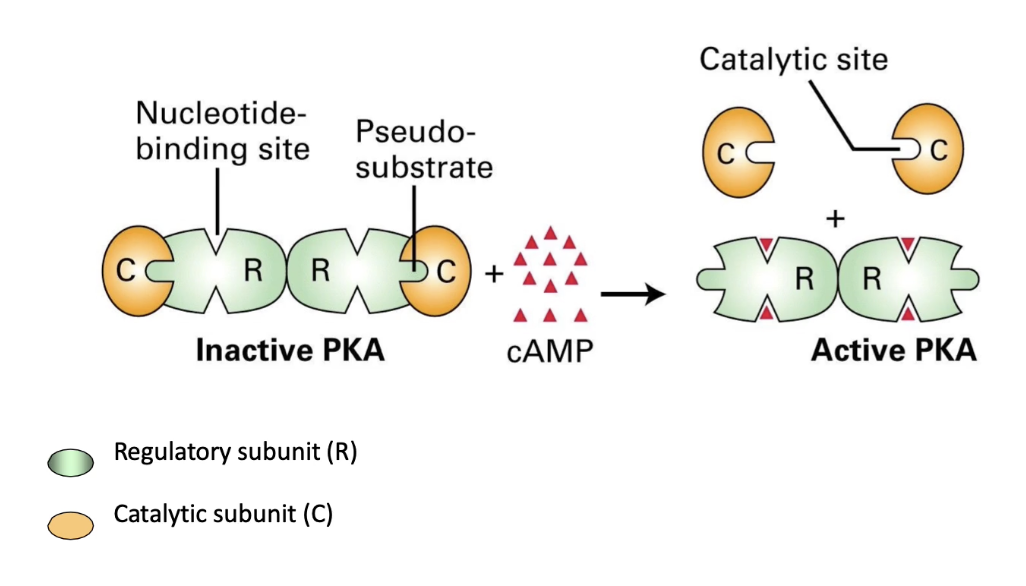
What role does the pseudo-substrate domain play in PKA regulation?
Pseudo-substrate domain (red) on regulatory subunit blocks catalytic site in inactive PKA
With cAMP bound: pseudo-substrate retracts, freeing catalytic subunit → PKA active
Without cAMP: pseudo-substrate extends, blocking substrate binding → PKA inactive
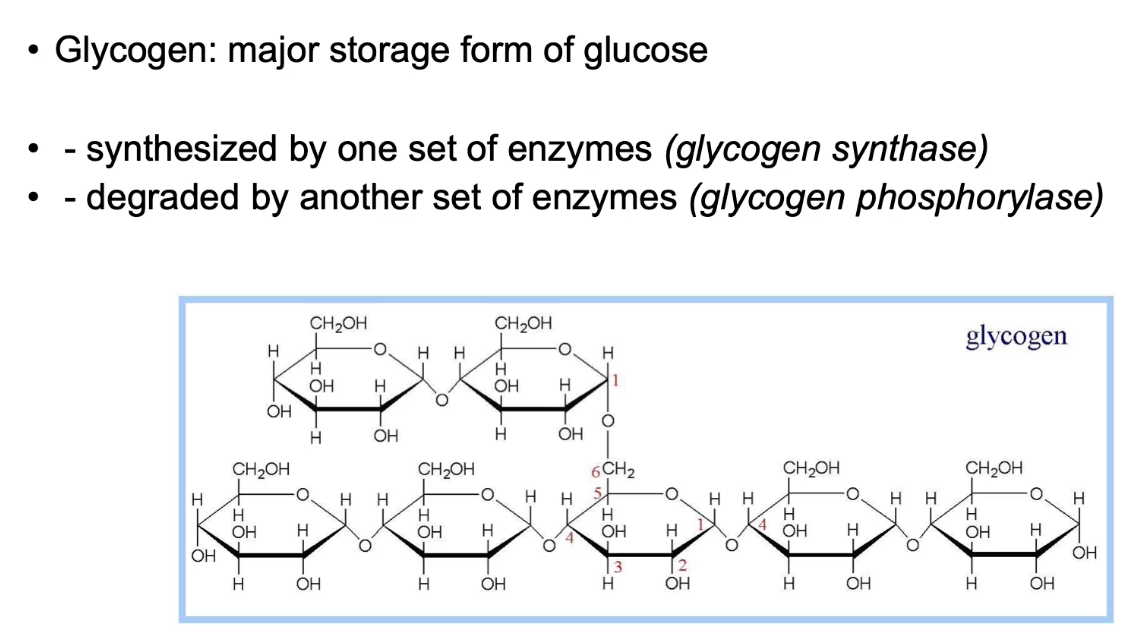
How does Protein Kinase A (PKA) regulate energy supply during the stress response?
PKA increases glucose supply by regulating glycogen metabolism
Inhibits glycogen synthase (stops glycogen formation)
Activates glycogen phosphorylase (promotes glycogen breakdown to glucose)
Released glucose feeds glycolysis → pyruvate + NADH → ATP production for energy during stress
How does PKA regulate glycogen metabolism differently in muscle and liver during the stress response?
Muscle:
Glycogen → glucose-6-phosphate → glycolysis → ATP for skeletal and cardiac muscle contraction
Liver:
PKA phosphorylates phosphorylase kinase → activates glycogen phosphorylase → glycogen breakdown
PKA phosphorylates/inactivates glycogen synthase → stops glycogen synthesis
Liver releases free glucose into bloodstream for other tissues
This is a fast, short-term response through enzyme modification, not gene expression
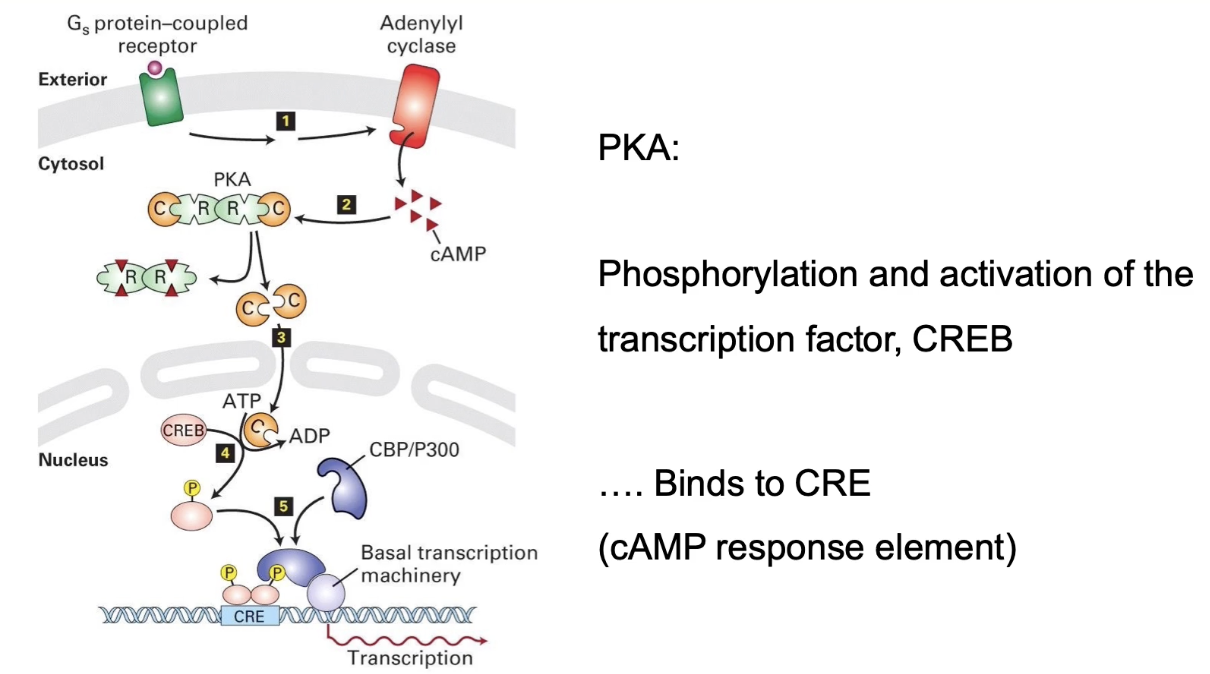
What is the long-term role of activated PKA in the nucleus during the stress response?
PKA catalytic subunit translocates to nucleus
Phosphorylates transcription factor CREB
CREB binds cAMP response element (CRE) on DNA
Initiates transcription of genes (e.g., phosphorylase kinase, glycogen phosphorylase)
Leads to increased production of enzymes for glucose production
Represents a slow, long-term response to stress signaling
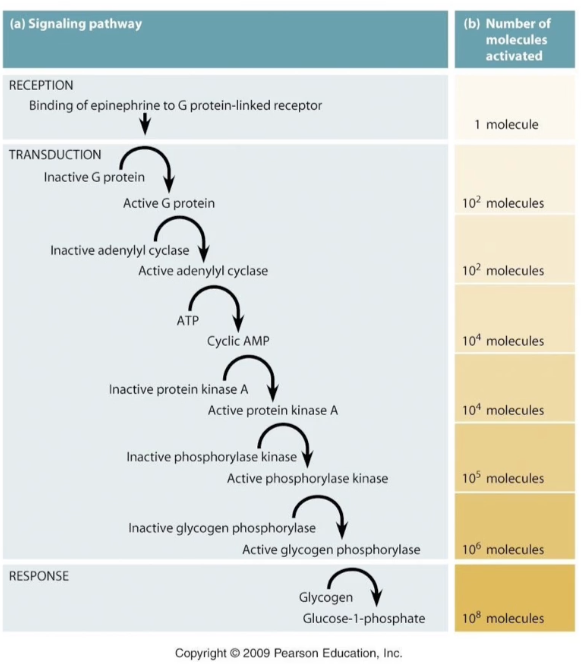
Why is signal amplification important in cell signaling pathways like the stress response?
Amplifies small external signals to large internal responses
Example: 10⁻¹⁰ M epinephrine triggers 10⁸-fold amplification
Amplification mainly occurs at enzyme activation steps
One adenylyl cyclase enzyme can produce ~100 cAMP molecules
Ensures sensitive and widespread cellular responses to low hormone levels
Applied Lecture
GPCRs and Disease
What are the key features of cell signaling via GPCRs, what types of signals exist, and which responses are fast vs. slow?
Receptor specificity: only cells with the matching receptor respond
Signal types:
Physical stimulus (e.g., light, touch)
Chemical molecules:
Small molecules (e.g., ions, gases)
Peptides or soluble proteins (e.g., hormones)
Proteins bound to cell surface or ECM
Signal transduction outcomes:
Fast responses:
Alteration of existing proteins (e.g., enzyme activation, second-messenger cascades)
Occur within seconds–minutes
Slow responses:
Changes in gene expression (e.g., transcription, new protein synthesis)
Occur over hours–days
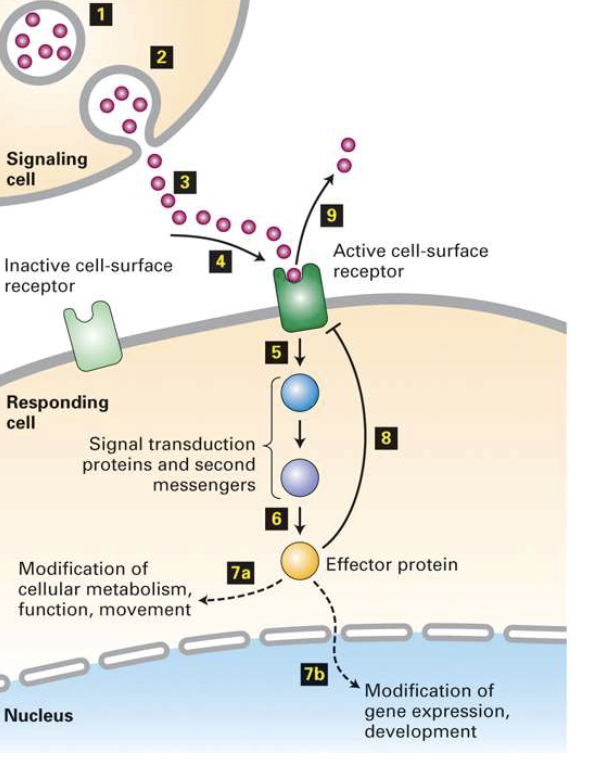
How does ligand binding to a receptor lead to a cellular response?
Receptor location: Found on or within target cells
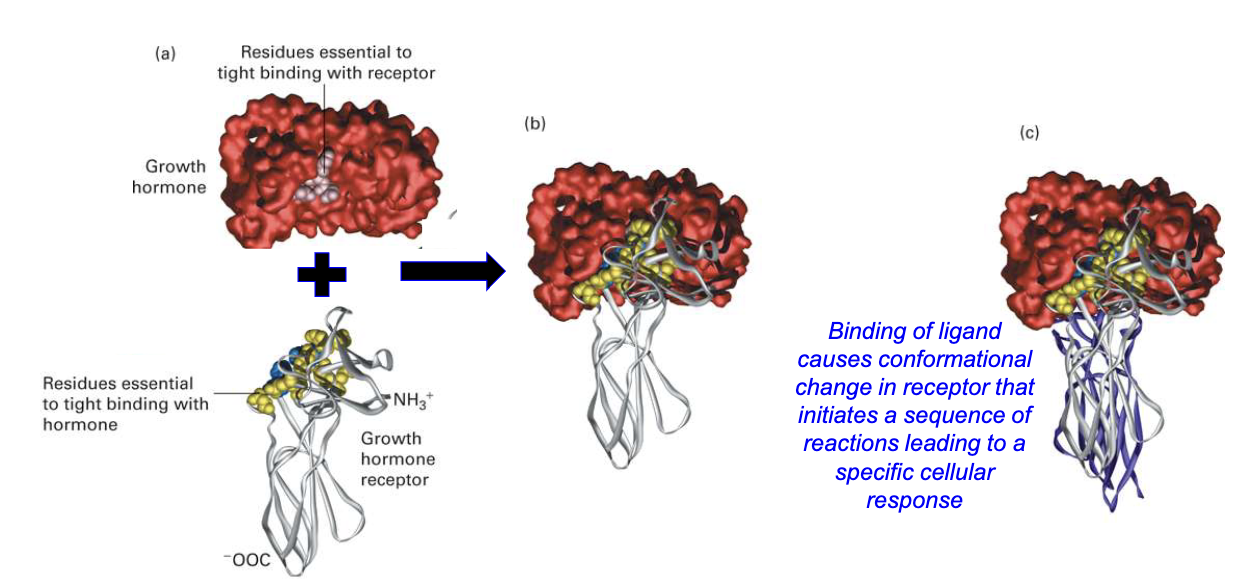
Specificity: Each receptor binds a specific ligand or related molecules
Binding mechanism: Weak, noncovalent interactions (ionic, van der Waals, hydrophobic) and structural complementarity
Effect of binding: Triggers a conformational change in the receptor
Outcome: Initiates a signaling cascade, leading to a specific cellular response
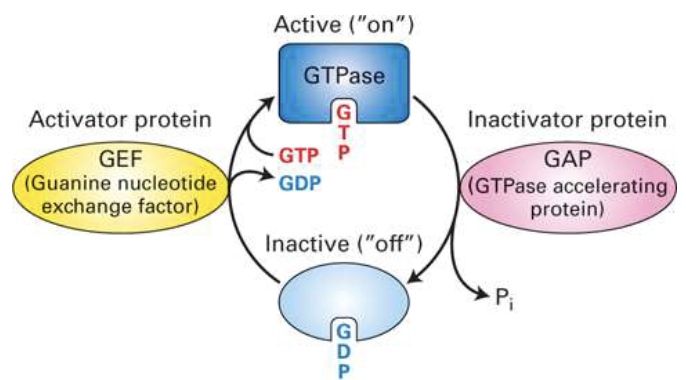
How do GTP-binding proteins function as on/off switches in signaling pathways?
GTP-binding proteins act as molecular switches that toggle between active and inactive states.
Activation: A GEF (Guanine nucleotide Exchange Factor) promotes exchange of GDP for GTP, turning the switch ON.
Inactivation: GTP hydrolysis (via intrinsic GTPase activity) converts GTP to GDP, turning the switch OFF.
Examples:
Trimeric G proteins (receptor-bound)
Monomeric G proteins like Ras (not receptor-bound)
What are intracellular second messengers and what is their role in signaling?
Ligands (first messengers) trigger changes in levels of low-molecular-weight second messengers
Second messengers are small molecules (e.g., Ca²⁺, cAMP, cGMP, DAG, IP₃)
They bind to proteins and alter their activity to propagate the signal
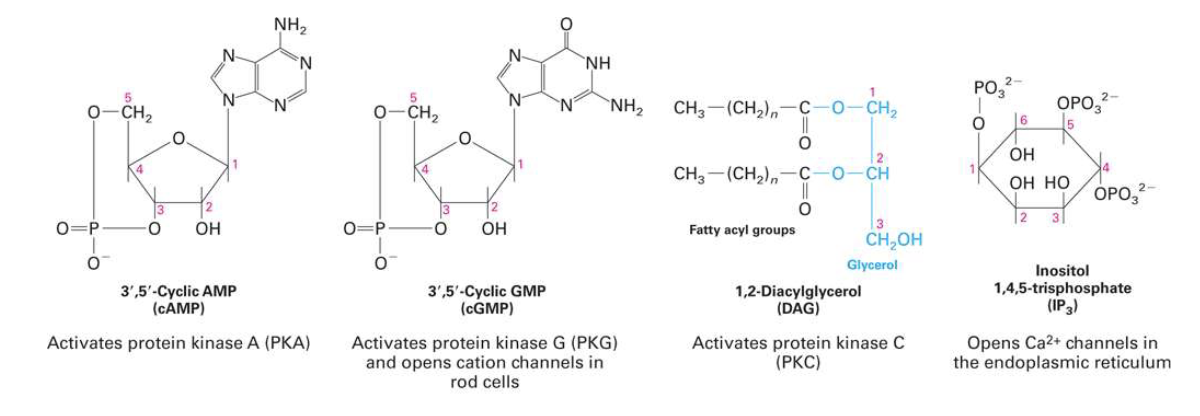
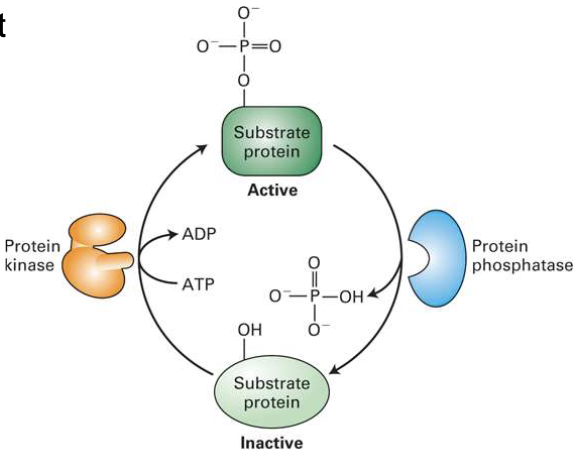
What is the role of protein kinases and phosphatases in cell signaling?
Kinases add phosphate groups (to serine, threonine, or tyrosine)
Can be cytosolic or receptor-associated
Phosphatases remove phosphate groups
Phosphorylation often enables protein-protein interactions
Human genome: ~600 kinases, ~100 phosphatases
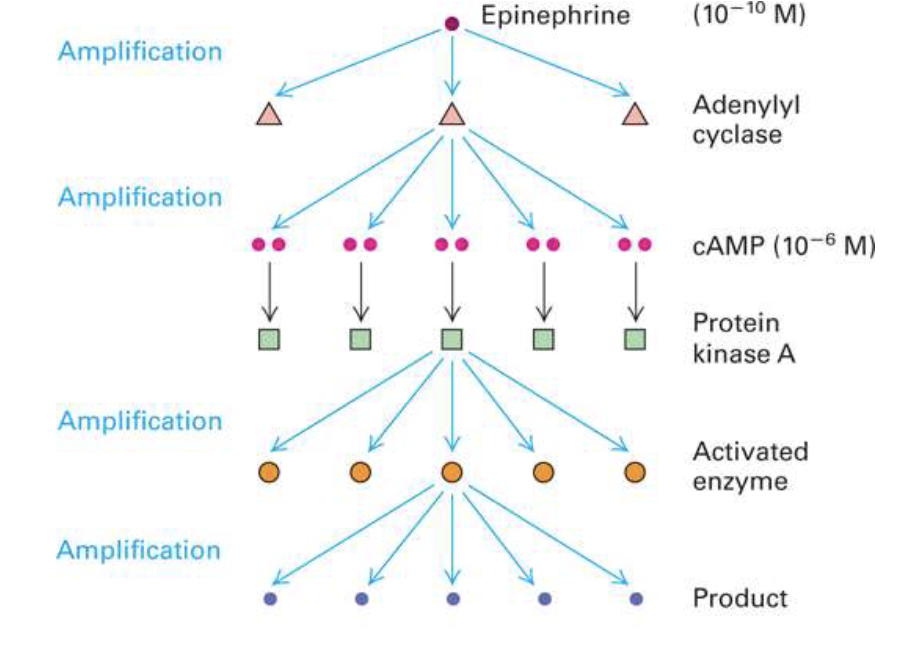
How does signal transduction enable signal amplification?
One ligand binding can activate a cascade
Each step can activate multiple downstream molecules
Results in hundreds of thousands of proteins activated from a single signal
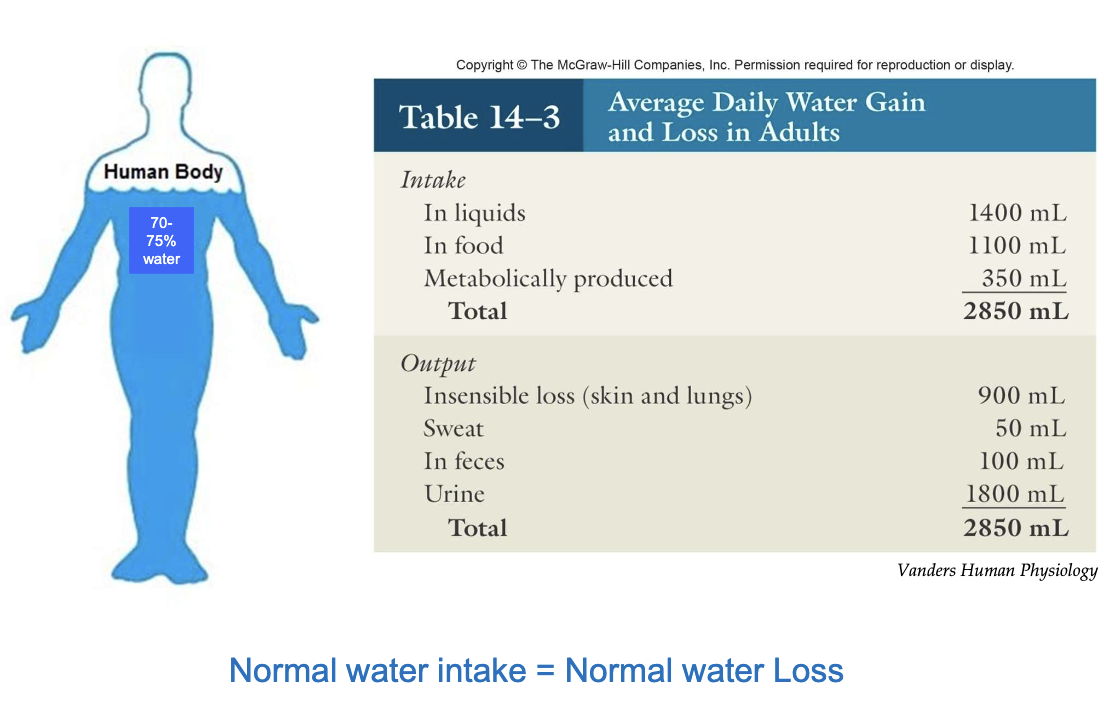
What maintains water homeostasis under normal conditions?
Output of water in our body comes from sensible (sweat, feces, urine) and insensible (skin and lungs) losses
Normal water intake is balanced by normal water loss
This balance maintains proper hydration and homeostasis
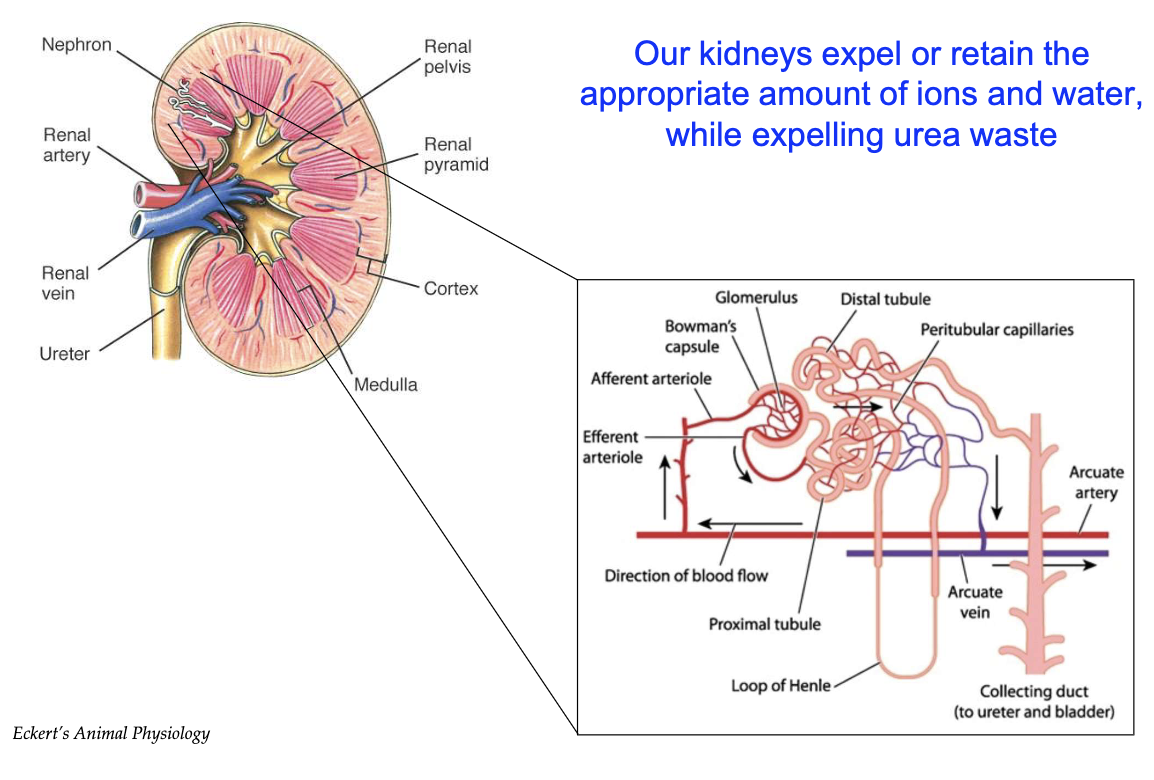
How do the kidneys maintain water balance and filter blood through nephrons?
Kidneys maintain water balance, crucial for cytoplasm, tissues, and blood plasma
Water loss occurs mainly via urine
Renal arteries bring blood to kidneys for filtration; renal veins return filtered blood
Nephron = functional unit of the kidney (millions in renal pyramids)
Blood flows: Renal artery → Afferent arteriole → Glomerulus → Bowman's capsule
Only small molecules (water, salts, ions, urea) pass into nephron; large ones (proteins, RBCs/WBCs) don’t
Fluid travels: Bowman's capsule → Proximal tubule → Loop of Henle → Distal tubule → Collecting duct
Reabsorption of water, salts, ions occurs along nephron via peritubular capillaries
Excess fluid becomes urine → bladder → excretion
Kidneys fine-tune reabsorption to maintain homeostasis
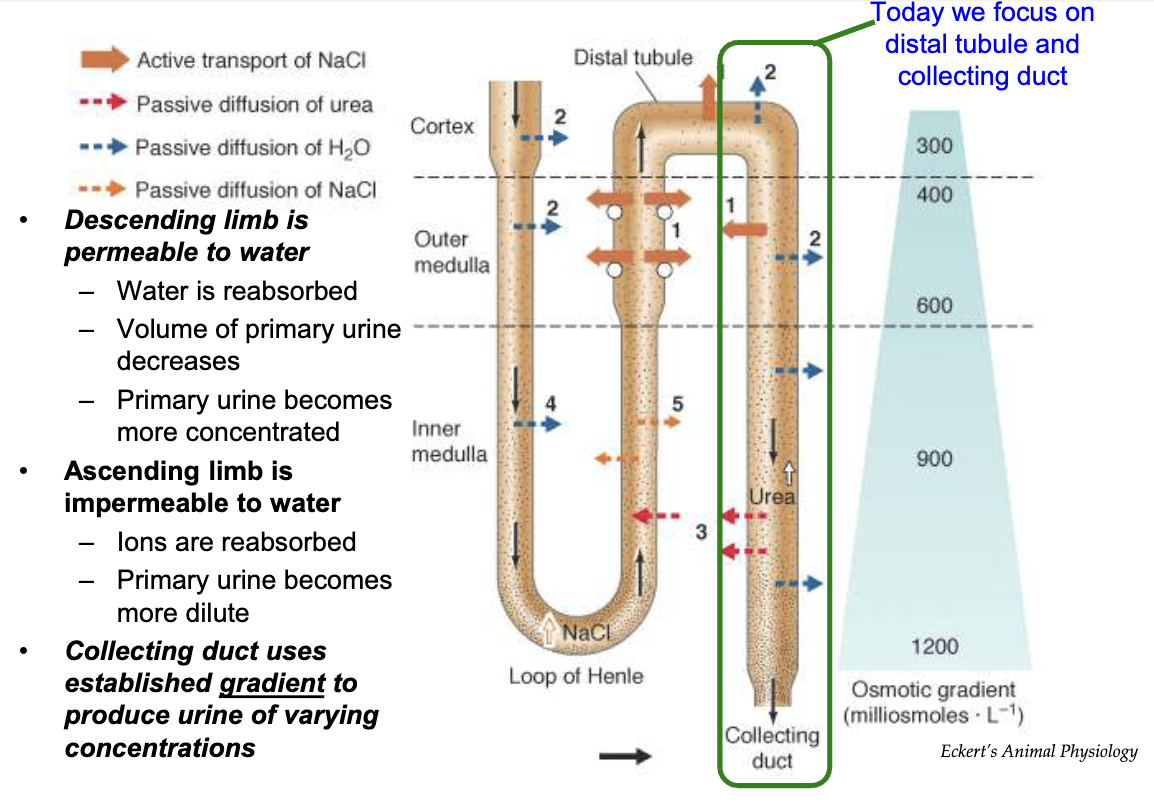
What happens in the descending and ascending limbs of the nephron during osmoregulation?
Descending limb of loop of Henle:
Water reabsorption via transporters
Water moves into interstitial space → peritubular capillaries
Urine volume decreases
Urine becomes more concentrated
Ascending limb of loop of Henle:
Impermeable to water
Ions reabsorbed (Na⁺, Cl⁻, K⁺)
Urine becomes more dilute
Collecting duct:
Final water reabsorption
Urine concentration increases
Remaining fluid → bladder → excretion
Uses gradient to adjust urine concentration
Osmotic gradient:
Increases from cortex to medulla in interstitial space
Drives passive water diffusion in descending limb & collecting duct
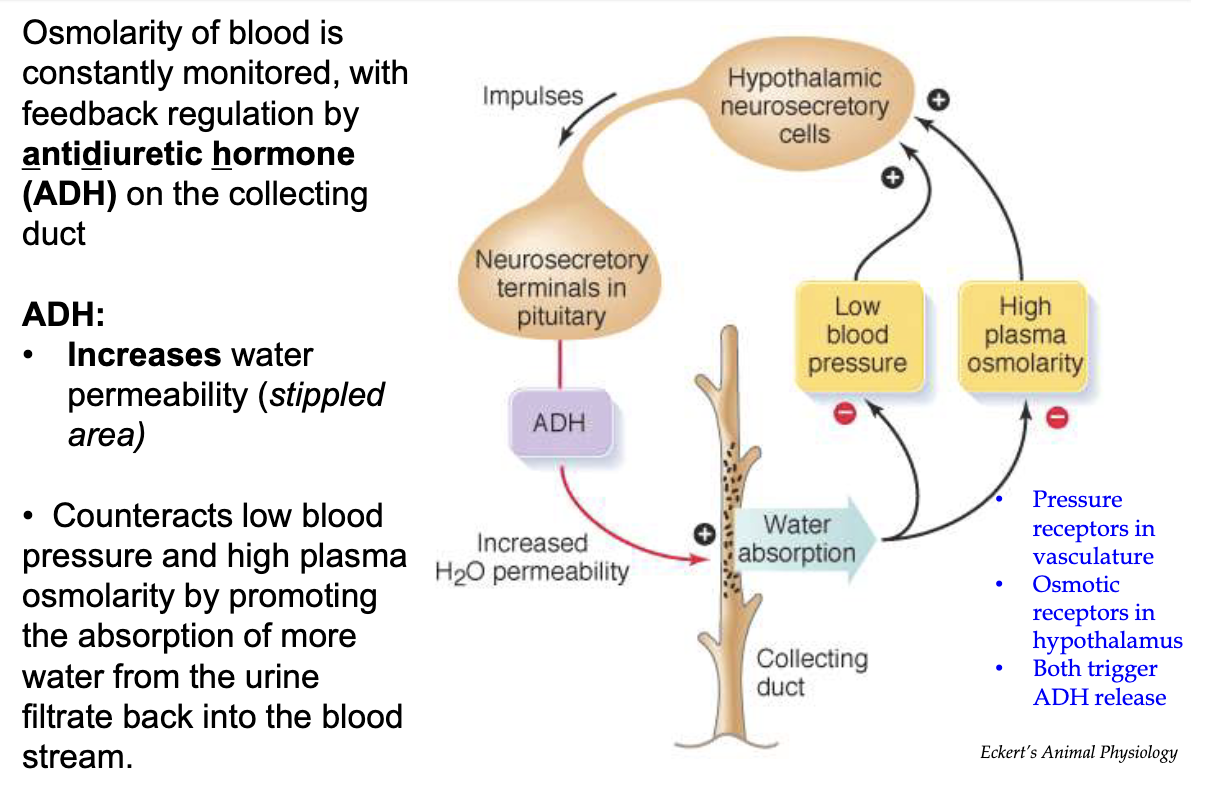
How does the body use ADH to respond to dehydration?
Trigger:
Low blood pressure
High plasma osmolarity
Sensors:
Pressure receptors in vasculature
Osmoreceptors in hypothalamus
Signal Pathway:
Hypothalamus signals posterior pituitary to release ADH
ADH Action:
Increases water reabsorption in collecting duct
Promotes water reabsorption from urine back into bloodstream
Helps restore blood pressure
Decreases plasma osmolarity
Mechanism:
Part of negative feedback loop to counter dehydration
What roles do ADH receptors and Aquaporin 2 channels play in the collecting duct?
ADH receptors (on basolateral membrane) bind ADH hormone
Trigger insertion of Aquaporin 2 channels on luminal membrane
Aquaporin 2 channels increase water permeability of principal cells
This allows water reabsorption from urine into the body
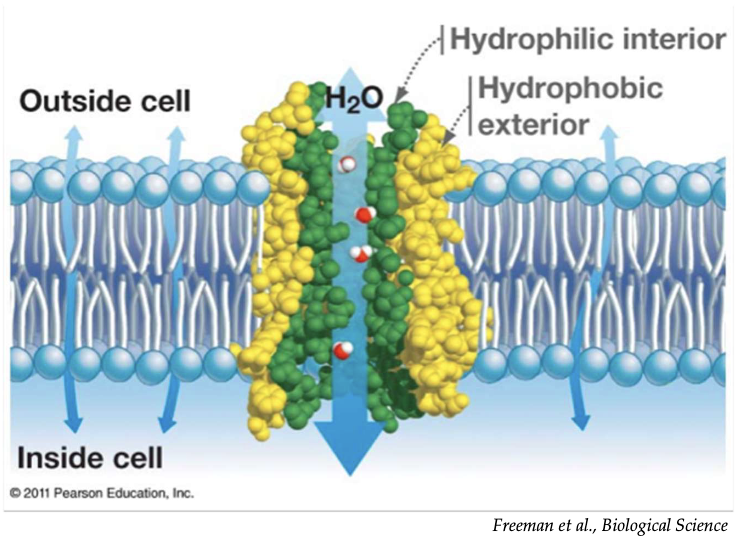
What is the function and selectivity of aquaporins in cell membranes?
Aquaporins are protein channels that allow rapid water transport
They are exclusively permeable to water (no ions or solutes)
Water moves through aquaporins by osmosis, following concentration gradients
How does ADH increase water permeability in the kidney's collecting duct?
ADH activates its receptor → activates Protein Kinase A (PKA)
PKA phosphorylates AQP-2 channels
Phosphorylation increases AQP-2 vesicle fusion to the luminal membrane and reduces endocytosis
Result: More AQP-2 channels are inserted and remain on luminal surface → increased water permeability
AQP-3 & AQP-4 are on basolateral membrane; AQP-2 constantly cycles to luminal membrane
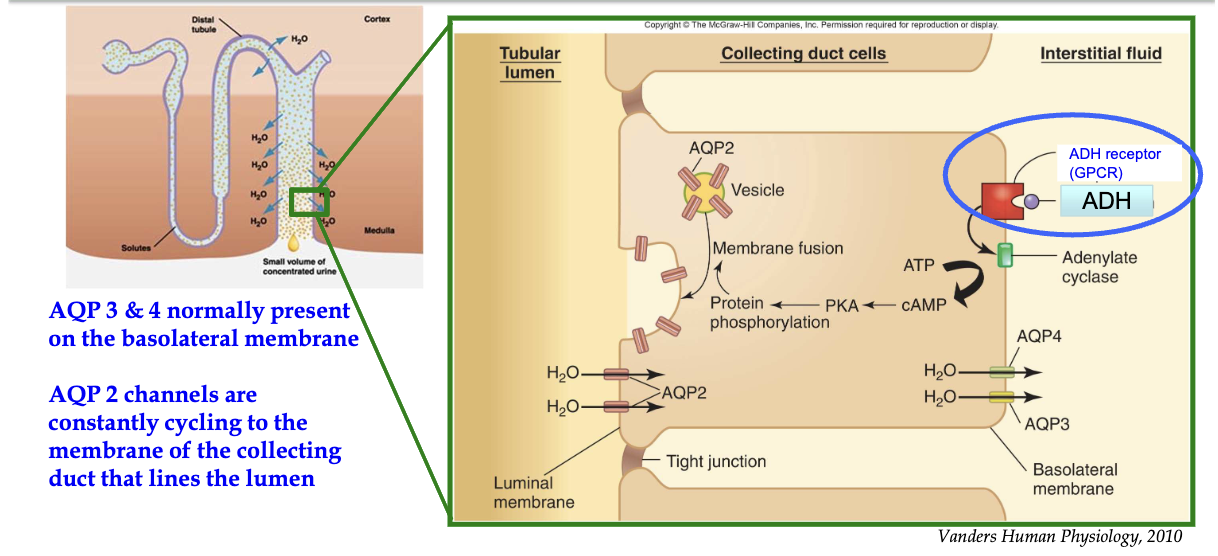
How does ADH regulate water reabsorption in the collecting duct via principal cells?
Trigger: Dehydration → low BP & high plasma osmolarity
ADH release: From posterior pituitary → enters circulation
Target: Principal cells in collecting duct (only cells w/ ADH receptors)
ADH binds: To GPCR on basolateral membrane
Signal cascade:
Activates adenylate cyclase → ↑ cAMP
Activates PKA
Leads to phosphorylation of proteins
Effect:
AQP2-containing vesicles fuse to luminal membrane
Water enters via AQP2, exits to interstitium via AQP3/4 (always present)
Regulation:
AQP2 insertion is ADH-dependent → prevents constant water reabsorption
Outcome:
Water reabsorbed into blood
↑ Blood pressure, ↓ plasma osmolarity
Negative feedback loop controls water homeostasis
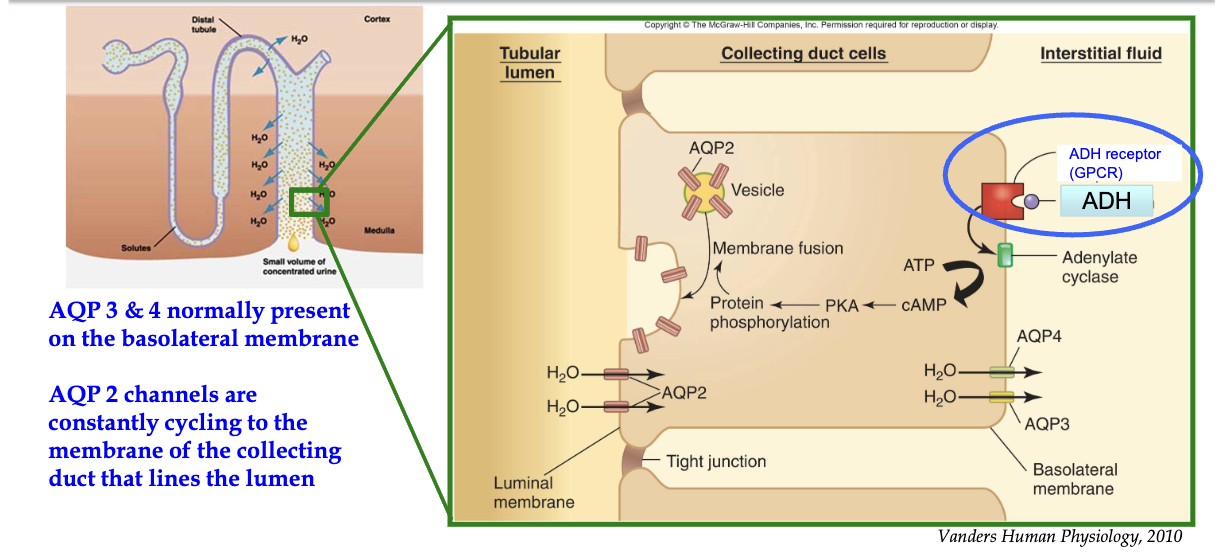
How do ADH receptors (GPCRs) regulate water reabsorption in the kidney collecting duct?
ADH binds to GPCR on basolateral (non-lumen) side → activates Gαs subunit
Gαs activates adenylyl cyclase → converts ATP to cAMP
cAMP activates Protein Kinase A (PKA)
PKA promotes exocytic insertion of AQP-2 channels to the luminal membrane
AQP-2 channels increase water reabsorption from collecting duct into blood

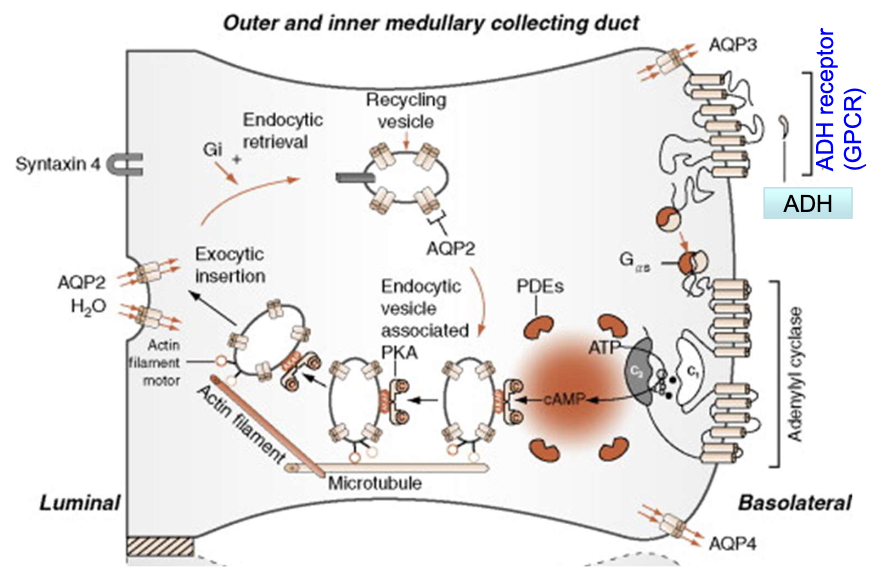
Describe the detailed signaling pathway activated by ADH in principal cells of the collecting duct.
ADH binds to GPCR (7 transmembrane receptor) on basolateral membrane of principal cells
GPCR functions as GEF (guanine nucleotide exchange factor)
Activates Gαs subunit → GDP swapped for GTP
Activated Gαs → stimulates adenylyl cyclase
Converts ATP → cyclic AMP (cAMP)
cAMP activates PKA
Binds to regulatory subunits → releases catalytic subunits
PKA phosphorylates proteins involved in vesicle trafficking
Promotes exocytic insertion of AQP2-containing vesicles into luminal membrane
AQP2 allows water to enter cell from nephron
AQP3 & AQP4 are always present on basolateral membrane → water exits into interstitial fluid
Inactivation of the pathway:
Phosphodiesterases (PDEs) degrade cAMP → reduces PKA activity
Without PKA, AQP2 insertion ceases
Outcome: Regulated water reabsorption based on ADH levels
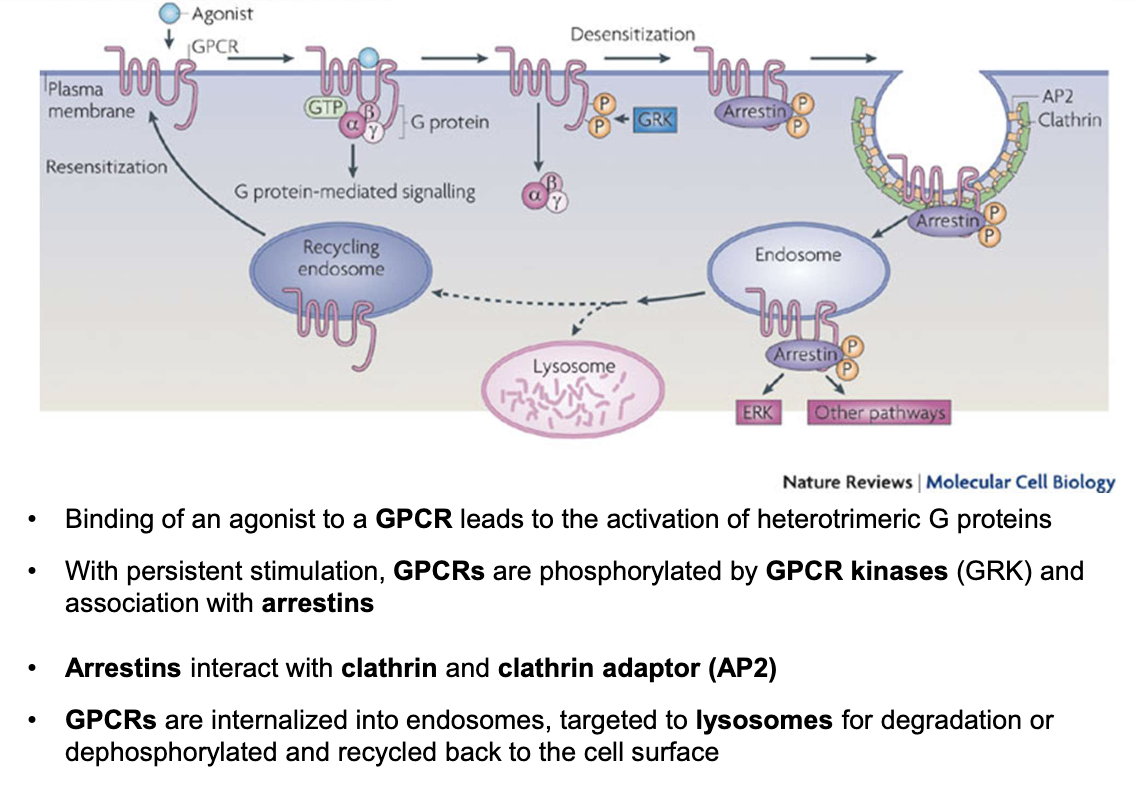
How is GPCR activity regulated after persistent stimulation?
Persistent agonist binding → GPCR phosphorylated by GPCR kinases (GRK)
Arrestins bind phosphorylated GPCRs
Arrestins recruit clathrin and adaptor protein AP2
GPCRs internalized into endosomes
GPCRs either degraded in lysosomes or dephosphorylated and recycled to the membrane
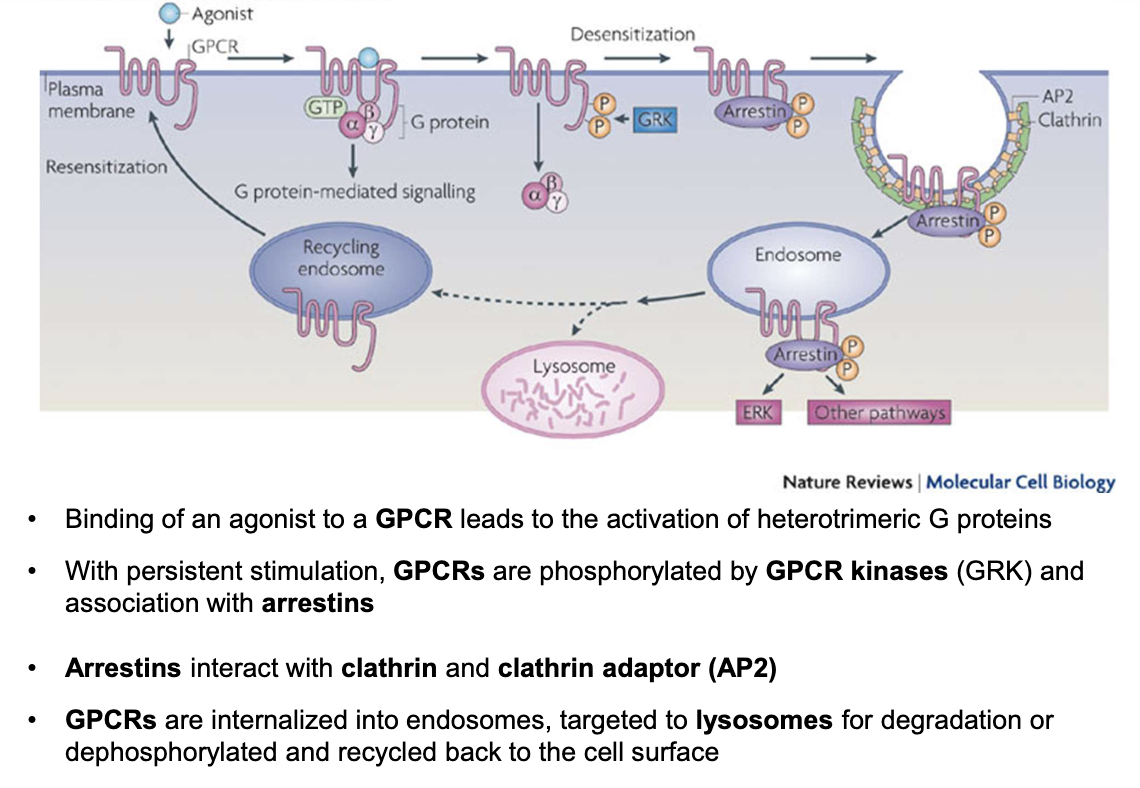
How is ADH GPCR signaling regulated at the basolateral membrane of principal cells?
GPCRs are not permanently present on the membrane; they undergo dynamic regulation
Agonist binding (e.g., ADH) → activates G-protein signaling
Persistent stimulation → receptor desensitization
Mediated by GPCR kinases (GRKs) → phosphorylation of GPCR
Phosphorylated GPCR binds to arrestin
Arrestin recruits clathrin & AP-2 adaptor protein
→ Leads to endocytosis of GPCR into an endosomeEndosome fate:
Lysosomal degradation (receptor destruction)
Dephosphorylation & recycling back to basolateral membrane
Net effect:
Controls sensitivity of cells to ADH
Regulates intensity and duration of water reabsorption response
What causes Congenital Nephrogenic Diabetes Insipidus (XNDI) and what are its main symptoms?
Caused by mutations in the ADH receptor (GPCR) — X-linked recessive inheritance
Mainly affects males; females are carriers
Symptoms: excessive thirst, large volume of hypotonic/dilute urine (>3L/day, <250 mmol/kg)
Leads to dehydration, fatigue, seizures due to electrolyte imbalance, enlarged bladder
Nephrons are insensitive to ADH → impaired water reabsorption

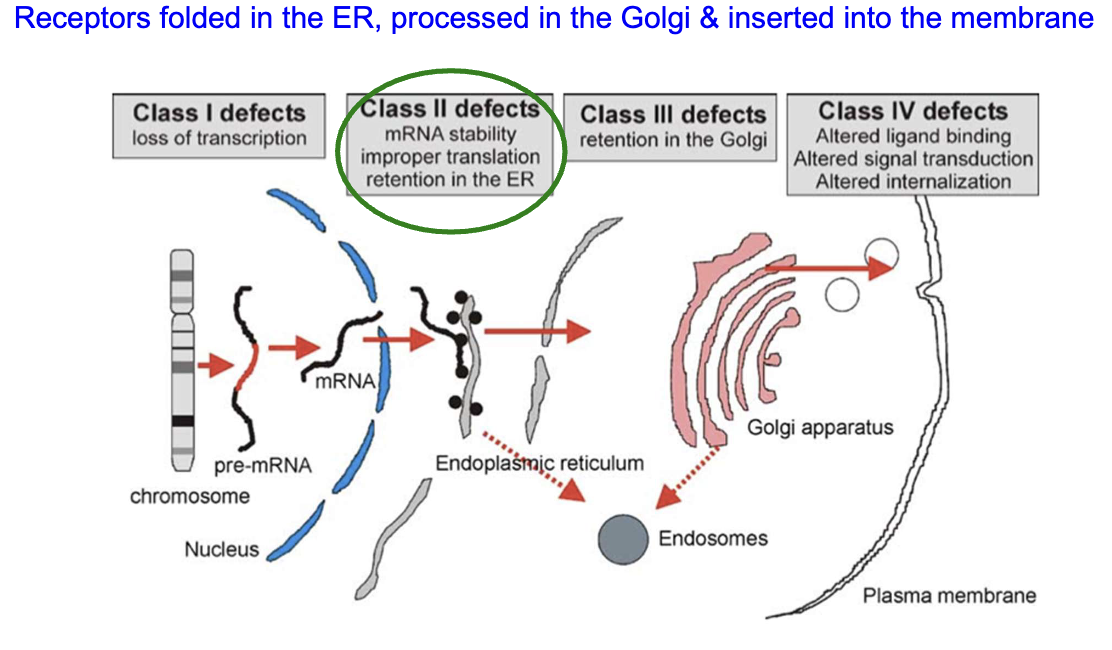
What are the four classes of GPCR defects and their main characteristics?
Class 1: Loss of transcription
Class 2: mRNA instability, improper translation, retention in the ER
Class 3: Retention in the Golgi
Class 4: Altered ligand binding, altered signal transduction, altered internalization
What causes nephrogenic diabetes insipidus (XNDI) at the molecular level?
Inactivating mutations (mostly Class II) in the ADH receptor gene
221 currently known ADH receptor gene mutations cause XNDI
Misfolded ADH receptors trapped in the endoplasmic reticulum
Loss of ADH signaling → no AQP2 expression or insertion into the luminal membrane
Results in impaired water reabsorption in the collecting duct
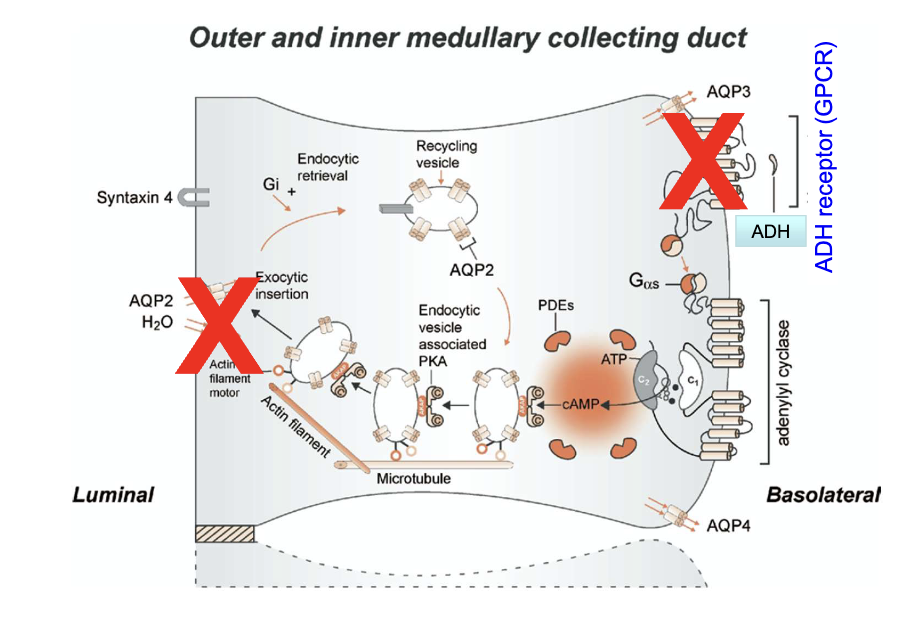
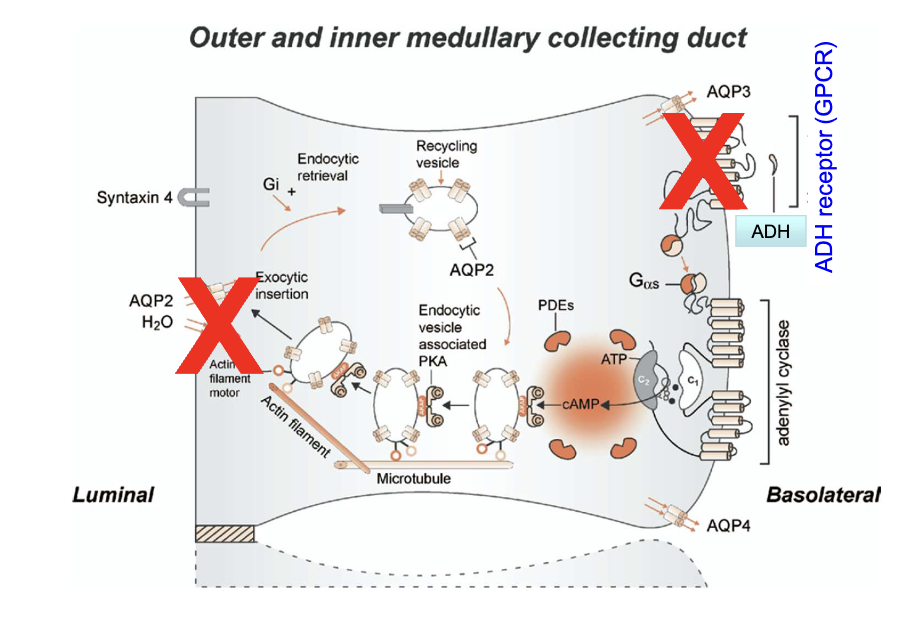
What is the mechanism of nephrogenic diabetes insipidus related to ADH receptor mutations?
Mutations cause absence of ADH receptors in the basolateral membrane
Leads to lack of aquaporin-2 (AQP2) channels in the luminal membrane
Result: impaired water reabsorption in the collecting duct causing excessive urine output, irritation and damage to urinary bladder
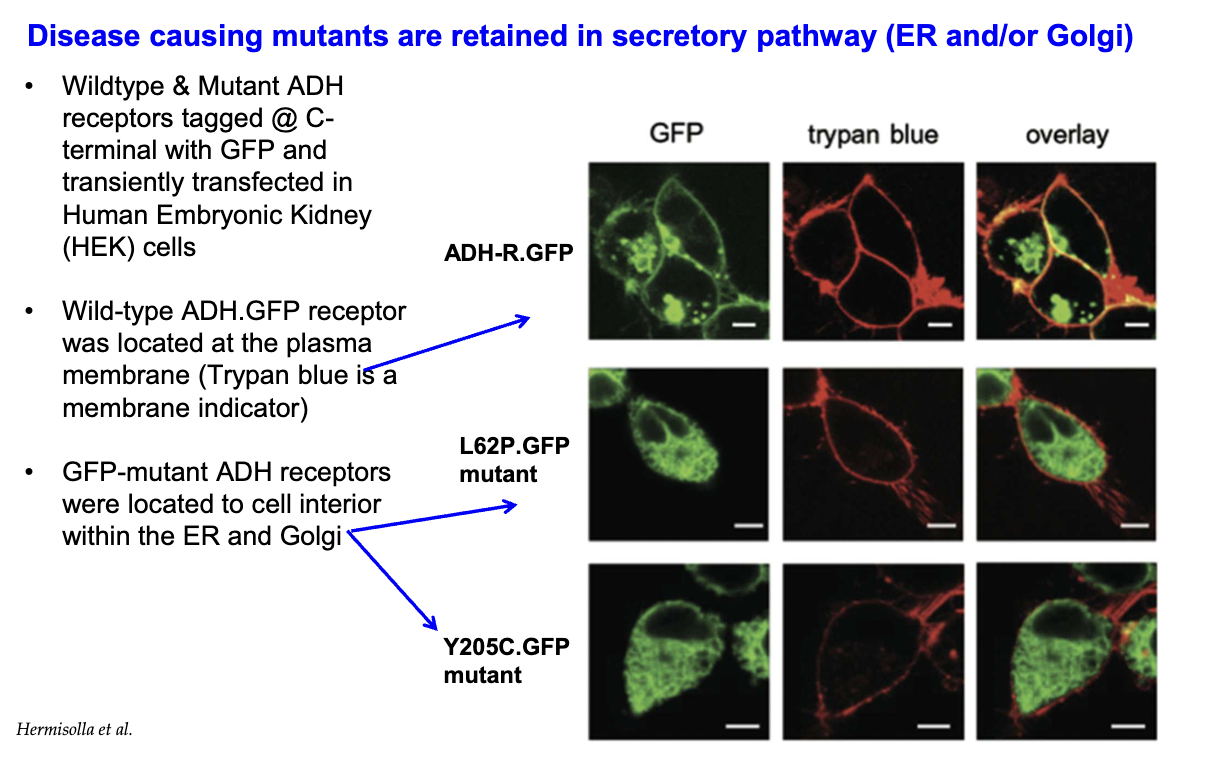
How do Class II ADH receptor mutants behave in cells compared to wild-type receptors?
Wild-type ADH receptors localize to the plasma membrane.
Class II mutant ADH receptors (e.g., L62P, Y205C) are retained inside the secretory pathway, trapped in the ER and Golgi.
This retention prevents receptor trafficking to the membrane, impairing function.
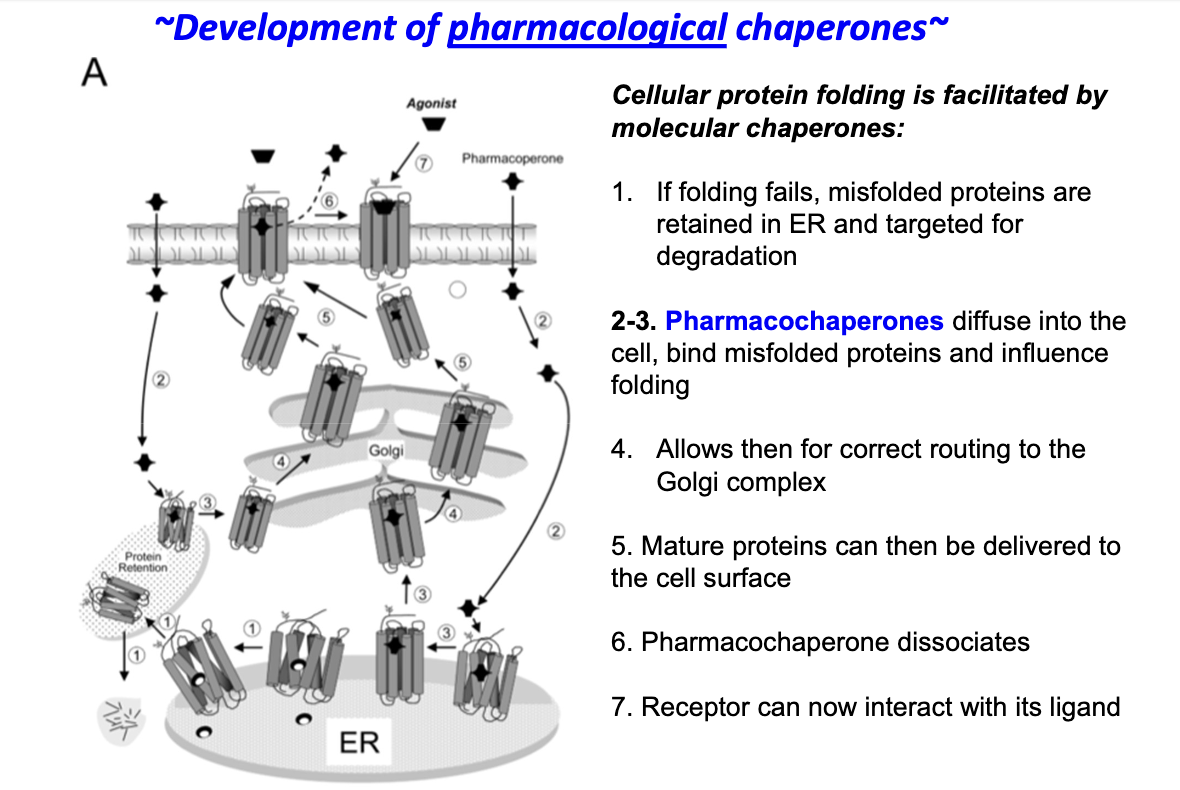
What are pharmacological chaperones and how can they help with Class II ADH receptor defects?
Synthetic small molecules that assist in proper protein folding
Bind target proteins via non-covalent interactions (van der Waals, hydrogen bonds)
Stabilize native protein structure and prevent aggregation
Allow misfolded ADH receptors to fold correctly and traffic properly in the cell
How do pharmacological chaperones help correct Class II ADH receptor folding defects?
Misfolded proteins are normally retained in ER and degraded
Pharmacochaperones enter the cell and bind misfolded proteins
They assist correct folding of proteins
Properly folded proteins are routed to the Golgi
Mature proteins reach the cell surface
Pharmacochaperones dissociate
Receptors can now bind their ligand and function
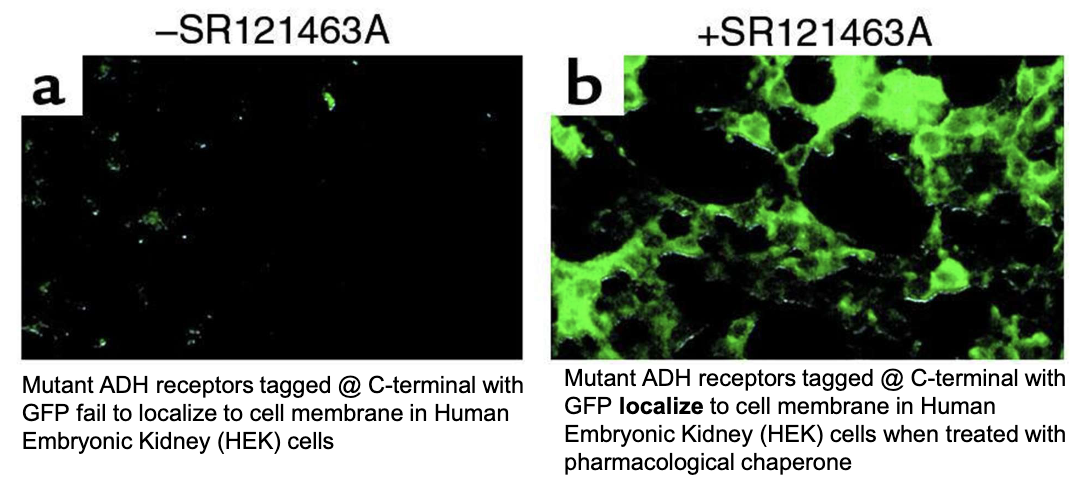
How can pharmacological chaperones like SR121463A help mutant ADH receptors in HEK (human embryonic kidney) cells?
Rescue receptors by:
Promote proper folding of mutant ADH receptors
Increase receptor localization to the cell membrane
Restore responsiveness to ADH
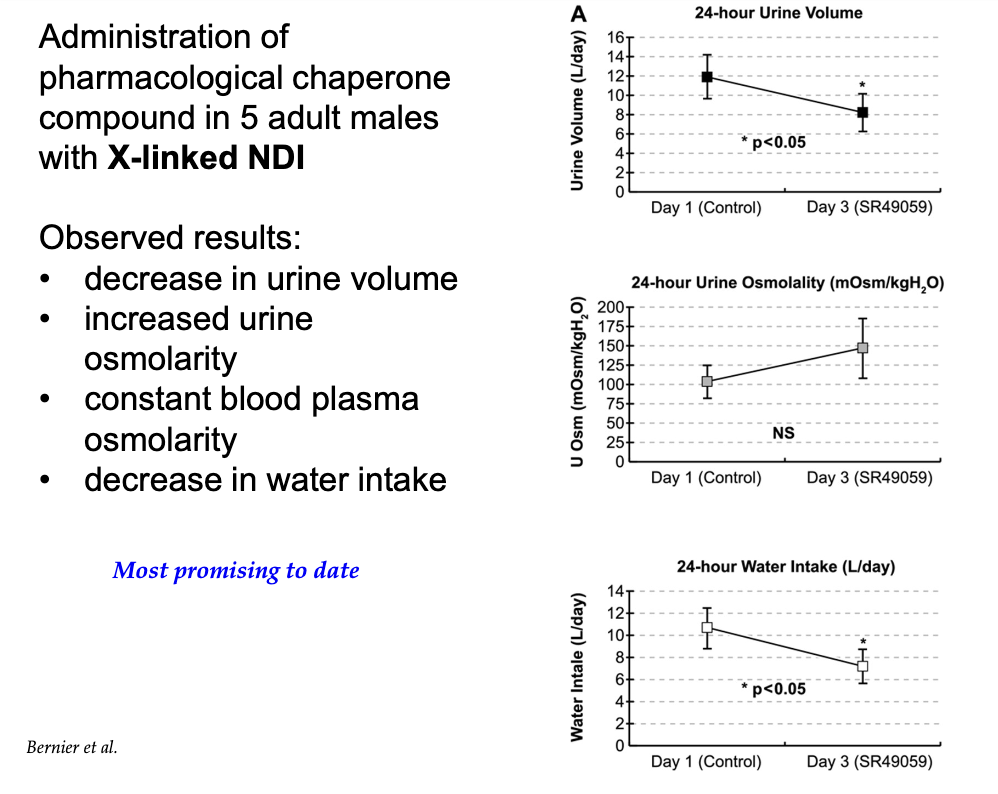
What effects were observed in adult males with X-linked nephrogenic diabetes insipidus after treatment with pharmacological chaperones?
Decreased urine volume
Increased urine osmolarity
Stable blood plasma osmolarity
Decreased water intake (less signals for thirst)
Most promising treatment to date.