Structure based drug design
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
29 Terms
Drug discovery pipeline
Usually would take 12 years between target protein and drugs that could be sold on the market, but recently, due to structure based drug design, this timeline collapsed down considerably
Drug design pipeline involves multiple phases:
Prediscovery
Trying to understand a protein in a system that may have sometherepeutic benefit
Understanding the basic mechanism behind disease
Designing assays to assess the system
Hit-to lead discovery
Once we’ve identified a target, potentially even a compound
Optimisation steps —> micromolar to nanomolar affinity
Preclinical studies
Toxicity tests in animals
Move on to clinical trials
Goal is to identify candidates early on and reduce the attrition process down

Streamlining this process
For optimising target identification and validation we can use software for modelling receptor structure
Accurate homology models which can be used to test the activity of various compounds
Screening in silico of high numbers of compounds with high degree of accuracy
Methods used in drug design
Biochemical assays
Thermal stability assays
Binding assays
Quantitative structure activity relationship
Computational
Docking
Molecular dynamics
Structure based
NMR
X ray crystallography
Experimental screening methods limitations
Need to be able to isolate the target molecule in large scale
Assays have to be optimised to screen a library of compounds
Analytical centrifugation for example can test up to 6 compunds and takes 2-3 hours
Microscale thermophoresis for example, can be done with 96 well or 384 well plates
However, the positive is that there are a diverse range of methods we can use —> some are ore or less amenable to high throughput analysis
Needs to be using ow levels of proteins otherwise it takes more money and effort for us to design more
Characterising Binding
These methods need to be able to tell us the Kd, the enthalpic and entropic contributions of the binding
We can then start to do structure activity relationship analysis which can help us to understand which parts of the protein are important in binding
Computational methods for design
We don’t need to express and purify the receptors in silico so advantageous
Compounds may be challenging to synthesise but can be made relatively quickly in silico and understand their properties
Can take the molecules of interest and use computational methods to assess binding sites
Determining affinity
To work out the Kd we must work out the delta G first which has an enthalpic and an entropic contribution
Trying to compute terms like entropy (more than entropy) and enthalpy can be challenging
Requires a structure or a good homology model and this can be difficult to determine the side chain conformation or the rotomer conformation
However, more recently, improvements in the quality of modelling has made this less of an issue
Molecular dynamics
Assumed that the chemistry is occurring under a classical regime (not 100% true) which allows you to assume Newton’s law and work out the forces that are acting on a particle
We know the mass of each particle so can work out the acceleration of the particles in response to the force
In a computer, we can then integrate this as a function of time
The main difficulty is working out the forces that are acting on the atoms —> we can do this by looking at the force field of each atom
separated into bonded and non-bonded interactions
Bonded interactions – the way that the covalent bonds, hydrogen bonds, steric clashing etc. react to being pushed part/pulled together
Non bonded interactions: electrostatic interactions – more challenging to deal with because you have to integrate across vast quantities of space but has been done
Why is this good for drug design
Molecules are free to behave as they want to behave —> can get them to adopt a different conformation etc.
We explicitly include the solvent (water molecules) and can understand about the entropic contribution
We have an atomistic description of the whole system and can understand how a particular drug interacts with a particular binding site
Challenges of molecular dynamics
Simulations nanoseconds in length but proteins bind in microseconds so can be hard to drag these trajectories out
Can take a long time for molecules to move from one site to another and means it takes a long time to sample different sites → so poor for determining where binding sites are if you don’t know but good if we insert the molecule into the binding site
Classical, not quantum technique so doesn’t deal with how the electrons move → if you have inhibitors which interact with the transporters/receptor this is not taken into account
If drugs react with pi-pi and pi-cation interactions can be difficult to capture the polarisation of the bonds in the force field that you’re using
Difficult to calculate the delta G of binding because we have water included in this system
However, this can be bypasses by calculating delta G, delta G in water and with the protein bound and because it’s a state function we can then calculate the delta G of binding
Docking
Makes the assumptions to simplify the calculation and find a mechanism to reduce the computational intensity so it can be applied to a large number of compounds:
Works off the basis that if you have a drug and a binding site, and the drug enters in the correct orientation, it can fit into the binding site → there’s a good chance if it fits in the binding site, it will be a good drug target
How do we identofy a binding site
Calculate all the possible conformations of the durg and calculate the energy of the resulting complex
Local
If you do a chemical labelling study using a native agonist and know that the binding site is in a particular location, we can screen for binding sites in a particular regons.
Then take all the conformations of the drug, rotate it in all the possible directions and see what the optimal interaction energy is
Systematic
If you don't have the information about the binding site can do a systematic search: create a box and move your protein around all different areas of the box at all different orientations
Calculate an interaction energy for each conformation and rotation and see what the optimal one is
Random:
Random conformations of the drug are placed randomly about the protein and the interaction is scored
Stimulated:
Molecular dynamic driven drug conformations, followed by stimulated annealing based docking
However, this doesn’t account for flexibility of the binding site or the protein and so can be refined using molecular dynamics to help define the structure
Scoring functions
Calculating interaction energy can be quite time-consuming, so we can use the same forcefield used for MD to calculate interaction energies
The downside is we don’t have any quantum effects (cation pi interactions, polarisation of bonds, charge effects)
Empirical or knowledge based approach
Faster, less computationally demanding approach which is more suited to screening large libraries, is an empirical or knowledge-based approach
We know that certain types of interactions are important and how the directionality impacts the binding affinity
Develop an empirical forcefield using observations from previous studies which we can use to screen drugs – gives us a scoring function: the likelihood of a molecule of interacting with that protein, rather than a Kd
Computationally more efficient, can screen larger libraries
Statistical analysis of pairwise distributions
Types of docking
Rigid body
Receptor and ligands which you assume have a fixed conformation and you assume binding between the two
Computationally very efficient only 6 degrees of freedom
Flexible
Ligands and proteins have a range of different states —> usually assume flexibility of the ligand and can extend this to certain regions of the protein
But this slows this down and we have to test the energetics of hundreds and thousands of different conformations but is more accurate
Example: nAchR
Knew the conformation of the drug so can make the binding site flexible
Use docking techniques to see how the receptor responds to the presence of the agonist in the binding site
Combining molecular docking and molecular dynamics
Benefits of combining these techniques – not usually independent from one another
Use molecular dynamics to generate ensembles of structures of receptor protein
Let a molecular dynamics trajectory run and see that structure changes as a function of time
Cluster analysis: structures with several key featyres associated, take these and use the docking type studies to screen against a whole range of different compounds and understand which compounds will bind best and which conformational state its likely to bind to
When do computational methods work well:
When the binding pockets are well defined
When there’s limited conformational flexibility in the drug and the target protein
Struggle with cryptic binding sites (e.g: sites of protein-protein interactions)
Easier to find the binding sites for rigid antagonists which do not induce a conformational change
Even if you don’t get an exact answer, can be used to:
Opens up new conformational spaces which you can study which you hadn’t previously considered
Can use it to design experimental screens if you know certain motifs favour binding
NMR based methods
Ligand based
Rely on observation of the ligand, not the protein
No requirement for labelling of the target which can be good if your protein expressed very poorly
Typically, data acquisition is fast
No size restriction of the molecule, so can quickly screen large numbers of compounds
Examples:
Saturation transfer difference NMR (STD-NMR)
Transferred NOE (trNOE)
Target based
Rely on looking on what the target of the ligand is
Need to label the protein with carbon-labelled isotopes
Expensive
Can then titrate in drugs and see how the structure is changing
Can do entire structure elucidation
Limited to smaller target molecules
Does provide very site-specific information about how the drug is interacting with the protein
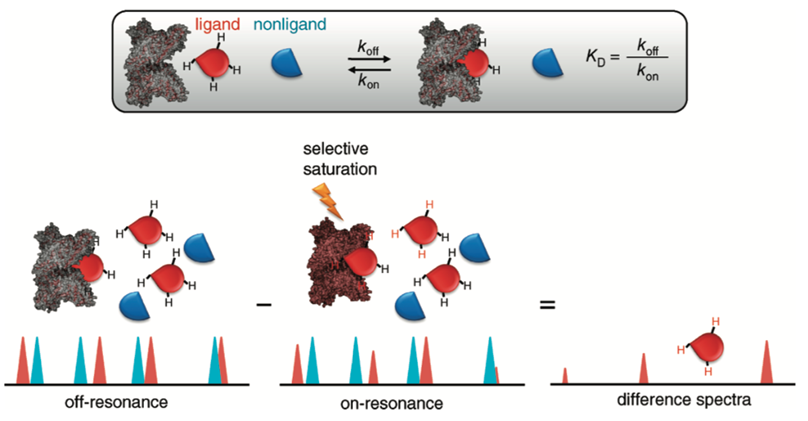
Saturation transfer difference (STD) NMR (ligand based)
The signal that you obtain for an experiment is based of the difference between the excited and the ground state
We can saturate - equally populate the ground and excited state to produce no signal
But we can selectively saturate resonances that come from the protein so the signal from the protein will disappear
If a small molecule binds to the target protein also becomes saturated to that the signal from the compound will become attenuated as it binds to the receptor
Attenuation of the signal can be used to identify which molecule is binding and so is amenable to screening a large number of proteins which can all be screened as a mixture
Example: HSA Binding
Tryptophan with a methyl group in 6 and 7 position
When you incubate these compounds, you find that the 7H3 binds and the signal is attenuated —> this protein is binding where the other one doesn’t

Limitations of STD NMR
Doesn’t give us a lot of insight into how the compounds are binding, so we go to protein-based screening
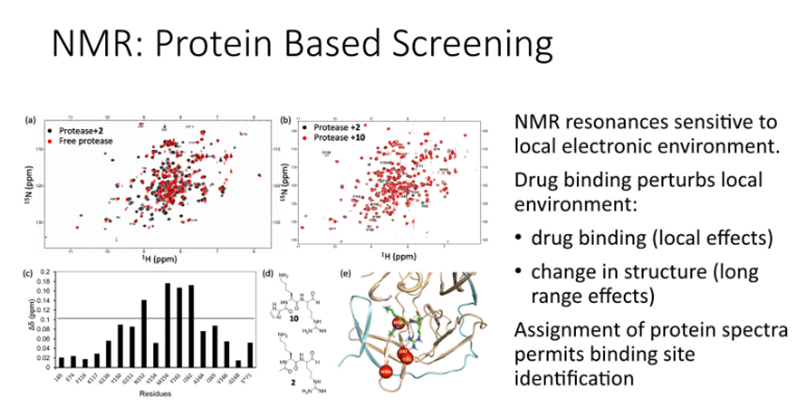
Protein based screening
HSQC style plots: every amide in the protein backbone gives rise to one point plotted à the position of the resonances in the spectrum is indicative of the protein conformation
If the drugs bind to the protein it changes conformations and the peaks move and look at how fast the peaks are moving
Enables us to measure a Kd and also allows us to identify which peaks have moved and identify a binding site of the protein
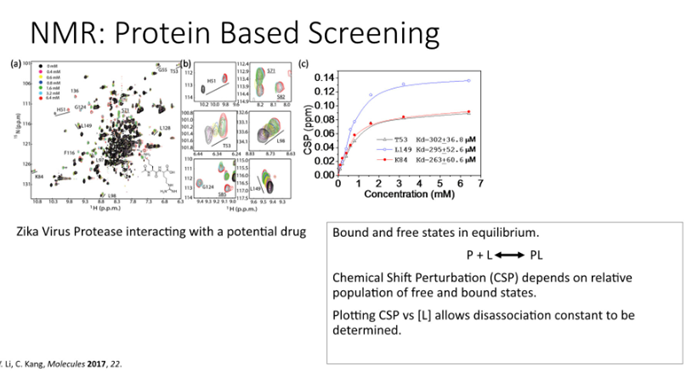
Example:
Titrate in protease from Zikka virus with a potential drug
When you titrate in inhibitor peaks move, can plot the chemical shift perturbation (movement of peaks as a function of concentration )
Fit these to models to work out the Kd
Crystallography
Use X ray crystallography to refine the structures and identify compounds
If we have a well-defined protein we can use high-throughput screening and fragment-based screening
High throughput screening: crystallise the protein in a range of different ligands —> see what binds in the binding site
A good set of diffraction data for compound with the target protein can be fast to resolve high resolution structural data which can be used to refine target compounds so you can improve the binding affinity
Feeds into a drug development cycle: can be used to design the next range of compounds with higher affinity until you get a clinical candidate
Drug target/structure – How do we get our drugs into crystal structures?
Cocrystallisation
Take a concentrated protein solution and add our drug of interest
If you leave this for a few days you will form a crystal of the drug bound to the target protein
Don’t need high solubility of drug
Protein ligand can be co-expressed
Requires screening of crystallisation conditions à conditions which gave you original structure of protein not going to be the ones which gives you a structure of protein and ligand
Relatively high throughput and are some techniques of automating it
Soaking
Take protein which you’ve already crystallised
50% of crystal is water/buffer àTake the crystal and put it in the buffer containing ligand of interest
Ligand will diffuse into the channels within the crystal and will bind to the binding site
Requires high concentrations of drugs so places limitations on the solubility of drugs
Assumes that the protein will not change upon ligand binding shape otherwise, the protein will fall apart
Fragment based drug design
Can generate chemical structures de novo
Not guided by a drug library
Take out target protein and incubate it with a fragment library – no longer looking for whole molecules binding woth high affinity
Small motifs which will bind t the drug and cause a shift in the thermal shift assay, NMR, or show some evidence of binding
Looking for any form of binding rather than high affinity
Can identify which fragments will bind and start to assemble them in a mechanism which will enhance the affinity of the target
If we link the two fragments:
The delta H doubled
The dela S is largely the same
Can begin to link the candidate drug —> slight decrease in conformational flexibility
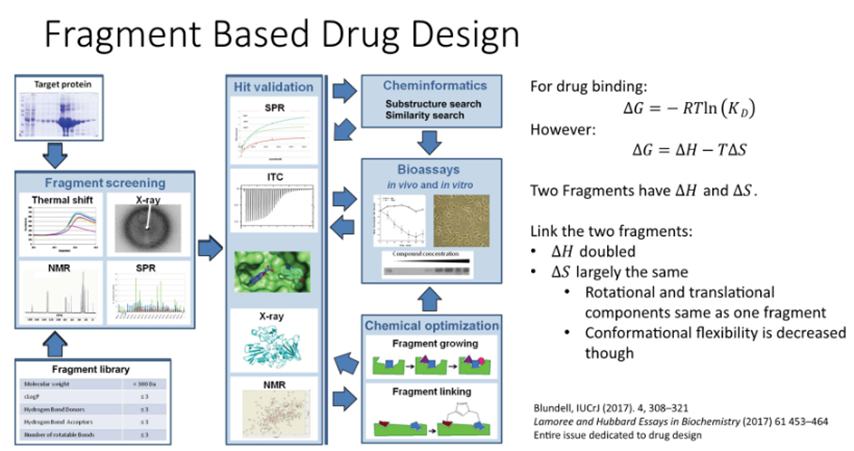
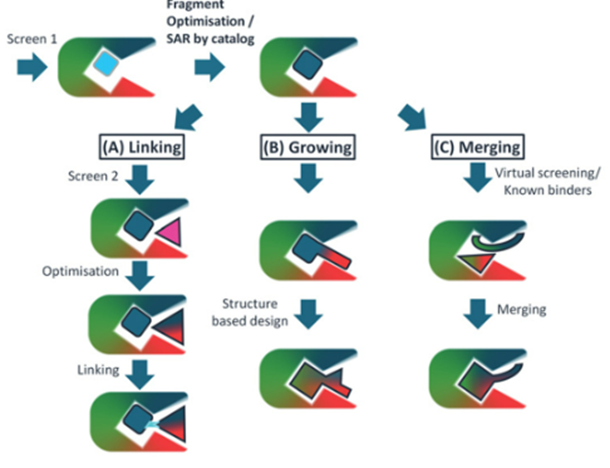
Strategies
Fragment optimisation
Bind multiple fragments and see which ones bind best
Linking
Linking and optimising of fragments
Growing
If we know the fragment binds well, we can look at binding it to other fragments to see if this improves
Merging
If 2 fragments which bind with high affinity, can look at merging these compounds in a way that they hold the same conformational space
Kd typically in the millimolar range which would not be effective, but by combining these fragments, we can reduce the Kd so that theyre in the nanomolar or micromolar range
Example: targeting SARS-Cov-2
Can create therapies which target the viral proteases or polymerase associated with the virus
Isolated the proteases and tried to identify compounds it was binding
X ray crystallography tried and screened a range of compounds
Found that the protease had a lot of water filled channels so tried soaking type experiments
Studies using fragment screens, mass spec: gave us 23 hits and 3 subsites
Ifentified binding to cys145 and therefore build a picture of what lind of molecules will inhibit the protease
Identify molecules which binds and target the catalytic triad which as a result targets the protein à Reactive N-chloro-acetyl group inhibits the protein
Then used fragments for selective inhibition
Example of a very rapid drug development which condensed the process down from 12-2 years