P3: PTMs, Complexomics and Peptidomics
1/25
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
26 Terms
PTMs
Challenges in analysing PTMs
Stoichiometry: for any given protein, a very small fraction will have PTM (in most cases).
Localisation: proteins can have more than one PTM type, for each type of PTM, a protein can have more that one (e.g. several phosphorylation on the same protein). It can be difficult to identify where the PTMs are on the protein, especially if they are close together.
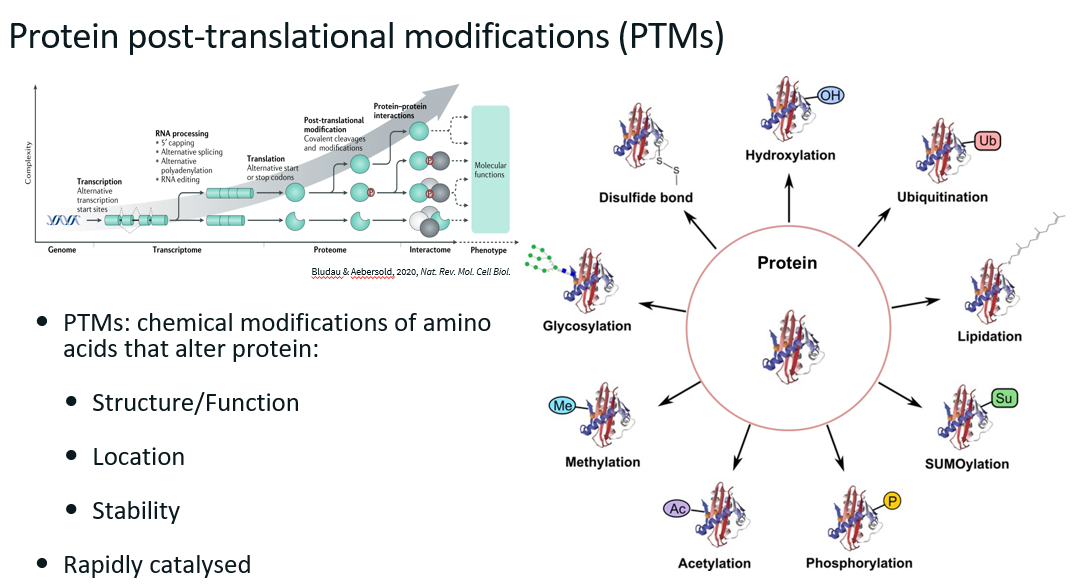
Main types of PTMs (6)
Phosphorylation
Acetylation
Methylation
Glycosylation
Ubiquitination
Sumoylation
Δmass indicates how the mass of the protein changes when modified by the corresponding PTM.

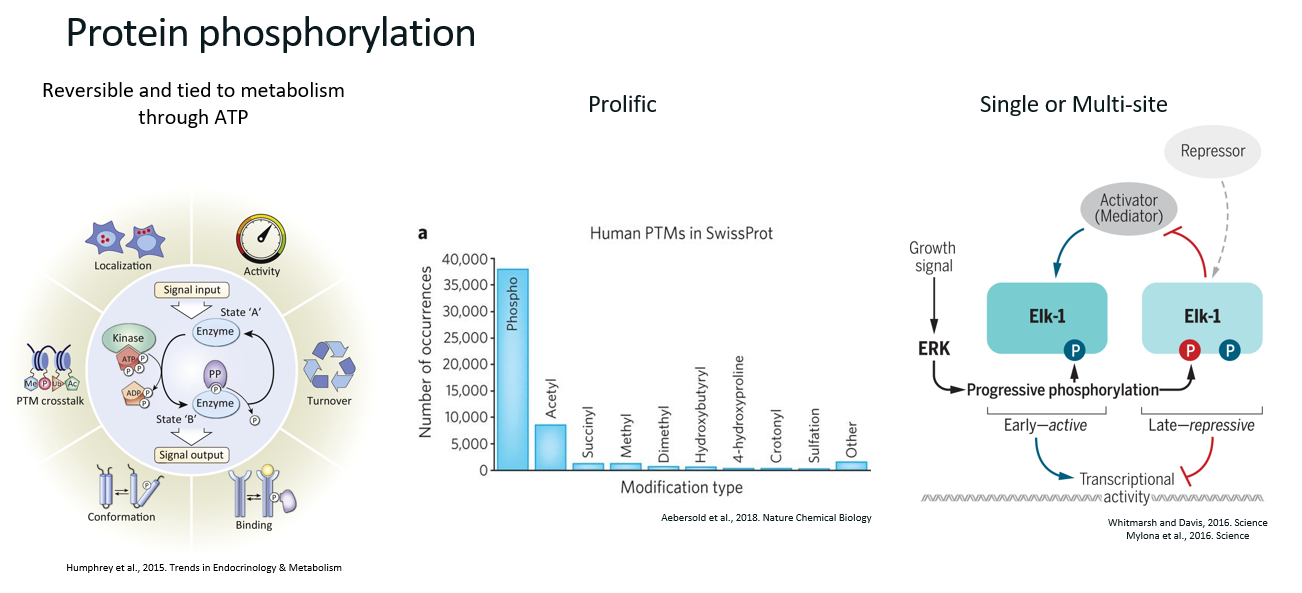
Phosphorylation (definition, main biological functions, characteristics)
Definition
Phosphorylation is the transfer of a phosphate group from a donor (often ATP) to an acceptor (e.g. a protein).
Main biological functions
Protein activity
Protein stability
Cellular location
Signalling pathways
Characteristics
Reversible
Prolific (the most common PTM)
Single or multi-site (i.e. proteins can be phosphorylated several times, which results in different functions).
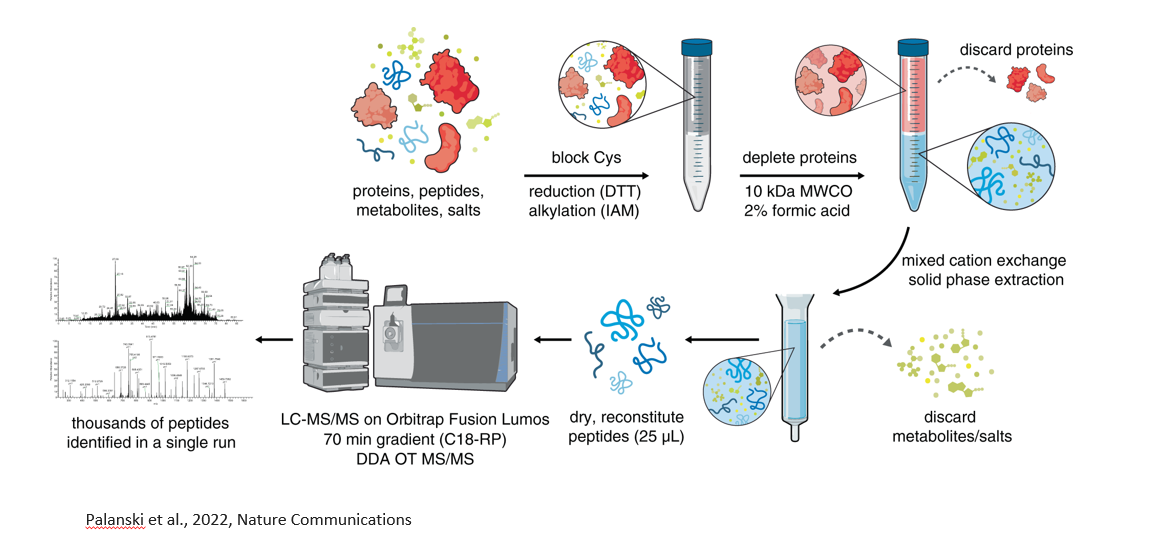
How do we circumvent the challenge of stoichiometry when studying PTMs?
For any given protein, a very small fraction will have PTM (in most cases). Thus, we need to get rid of the proteins that do not have PTMs and only keep the ones who do.
This is done by enrichment of the modified proteins. There are several techniques to do this based on the PTM.
Technique to enrich phosphorylated proteins
Phosphoenrichment
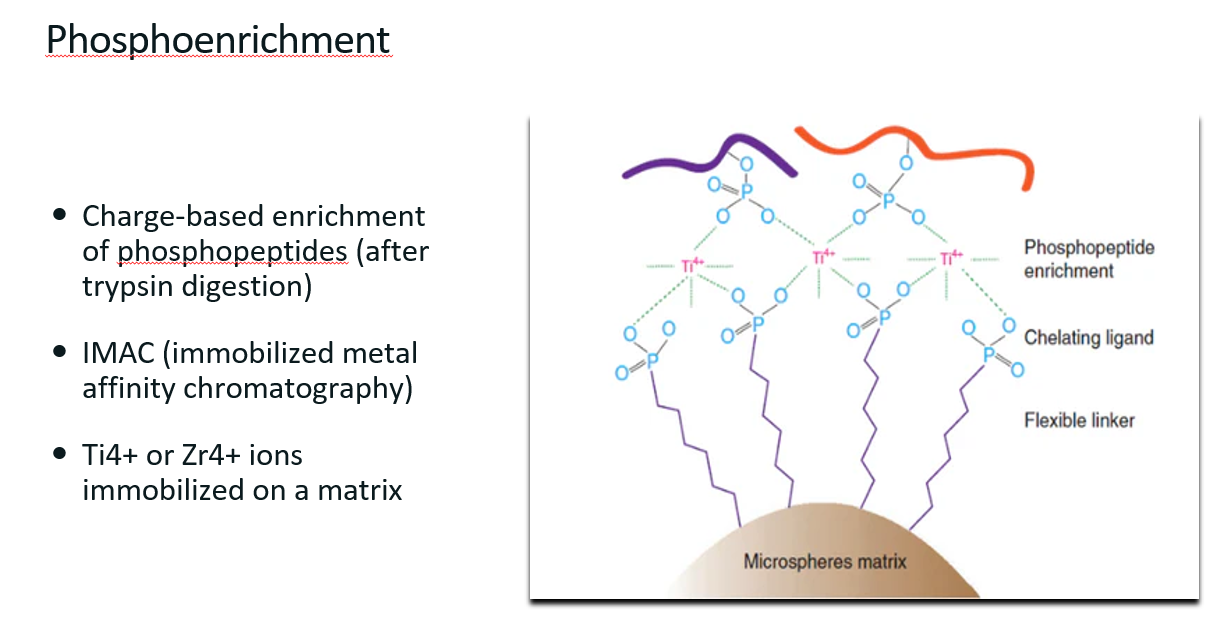
What is phosphoenrichment? (definition main technique used to do it)
Definition
Phosphoenrichment refers to a set of techniques used to isolate and enrich phosphorylated proteins or peptides from complex biological mixtures before mass spectrometry (MS) analysis.
This is a relatively easy thing to do since phosphate groups add a negative charge to the protein, a property that can be used to separate the modified proteins from the non-modified ones.
Immobilized Metal Affinity Chromatography (IMAC)
Charged-based enrichment of phosphopeptides (after trypsin digestion)
Uses metal ions (Ti4+ or Zr4+) immobilized on a bead.
These ions can bind to the phosphate groups to “pull out” the phosphorylated proteins from the sample.
Workflow in a Phosphoproteomics Experiment
Protein extraction from cells or tissues.
Proteolytic digestion, usually with trypsin, to generate peptides.
Phosphoenrichment to isolate phosphorylated peptides.
Mass spectrometry analysis to identify and quantify phosphopeptides.
Ubiquitination (definition, main biological functions, ubiquitination process)
Definition
Post-translational modification (PTM) where a small regulatory protein called ubiquitin is covalently attached to a target protein.
Ubiquitin is a small, 76-amino-acid protein that is highly conserved across eukaryotes.
Main biological functions
Protein degradation
Trafficking
Regulation of enzymes
Translation
DNA repair
Ubiquitination process
Ubiquitination is an enzyme-catalyzed cascade involving three main types of enzymes:
E1, Ubiquitin-activating enzyme: Activates ubiquitin in an ATP-dependent manner.
E2, Ubiquitin-conjugating enzyme: Transfers activated ubiquitin from E1 to E3.
E3, Ubiquitin ligase: Transfers ubiquitin to the substrate protein.
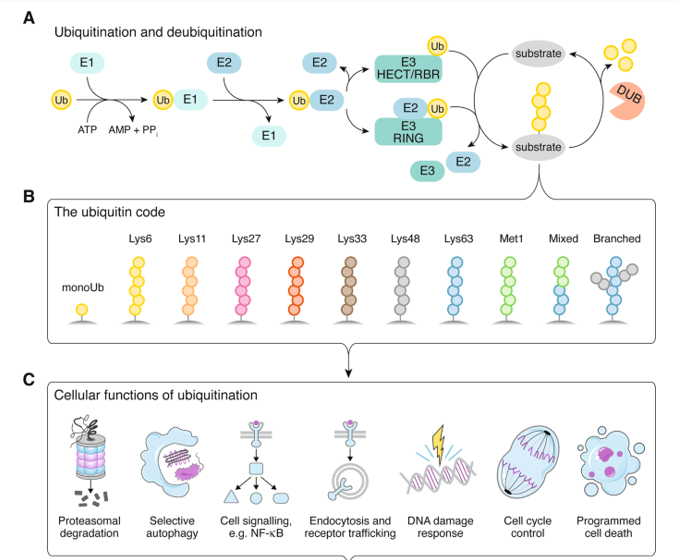
Diglycine tag
The di-glycine (diGly) tag is a signature remnant used in mass spectrometry-based proteomics to detect ubiquitinated lysines on proteins.
When a ubiquitinated protein is digested with trypsin, the ubiquitin itself is also cleaved, except for a small portion.
Trypsin cleaves at the C-terminal side of lysine (K) and arginine (R), but in the case of ubiquitin, the C-terminal Gly–Gly (G–G) motif that was attached to the lysine on the target protein remains bound to the lysine side chain. This leaves behind a di-glycine (Gly–Gly) tag on the lysine that was ubiquitinated.
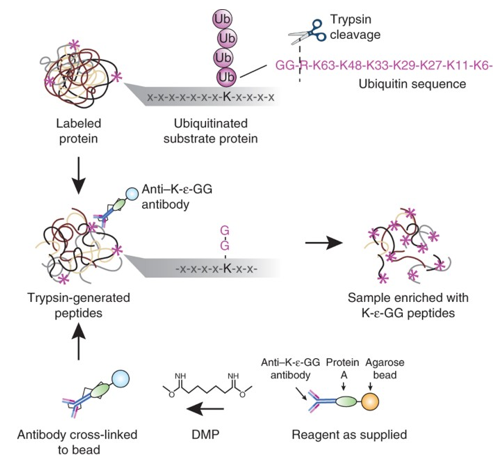
How to enrich ubiquitinated proteins?
After proteolytic digestion (usually with trypsin), peptides containing ubiquitinated lysines retain a di-glycine tag.
Specialized antibodies (anti-K-ε-GG) can be used to recognize and bind to this tag.
Workflow
Protein extraction from cells or tissues.
Trypsin digestion → generates diGly-tagged peptides at former ubiquitination sites.
Incubation with anti-K-ε-GG antibodies (usually coupled to magnetic or agarose beads).
Wash to remove non-specific peptides.
Elution of enriched diGly peptides.
Mass spectrometry (LC-MS/MS) to identify and localize ubiquitination sites.
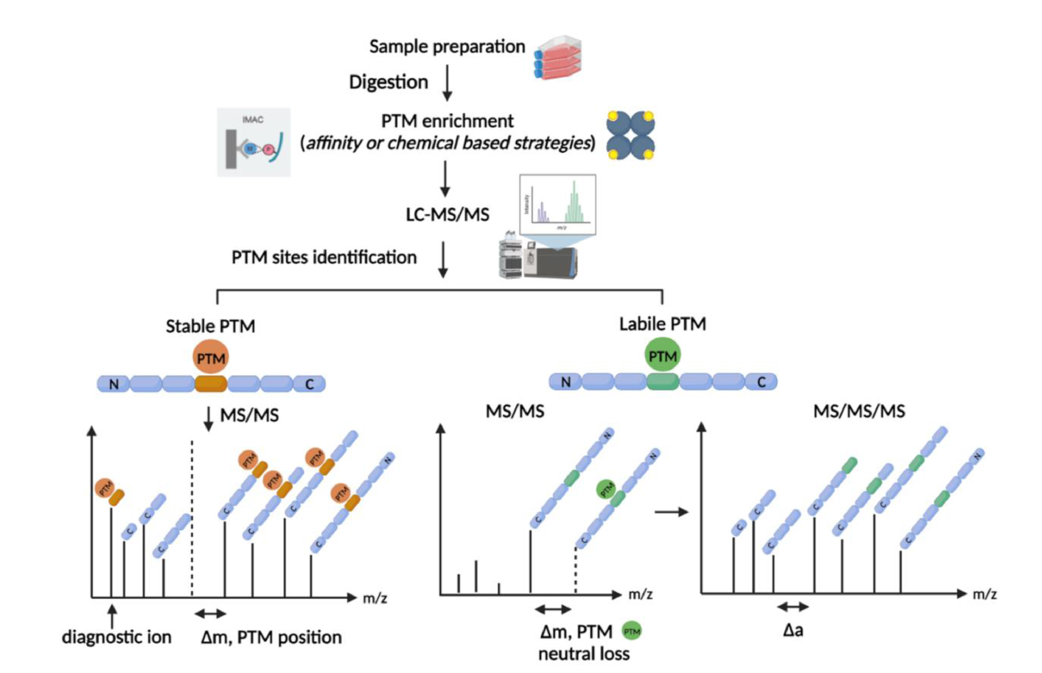
Labile PTMs
Labile post-translational modifications (PTMs) are modifications that are susceptible to fragmentation during mass spectrometry (MS) experiments.
What fragmentation method is used in Phosphoproteomics and why?
Reminder: In MS, fragmentation refers to the intentional breaking of peptide or molecule ions into fragments so their sequence can be determined. It's a core step in tandem mass spectrometry (MS/MS or MS²).
Phosphorylation is a labile PTM. Thus, in phosphoproteomics, ETD (Electron transfer Dissociation) is used over CID (Collision-Induced Dissociation) because it is more precise and preserves the phosphate groups. In CID, it is easy to loose the phosphate groups from the ions due to the brute-force of the technique.
General PTMomics workflow
Complexomics
Mass spectrometry-based method used to study macromolecular complexes.
Protein interactions are mediated by …
PTMs
Techniques to study protein interactions
Yeast two-hybrid screens
Affinity purification coupled to mass spectrometry
Proximity labelling
Cross-linking coupled to mass spectrometry
Co-fractionation coupled to mass spectrometry
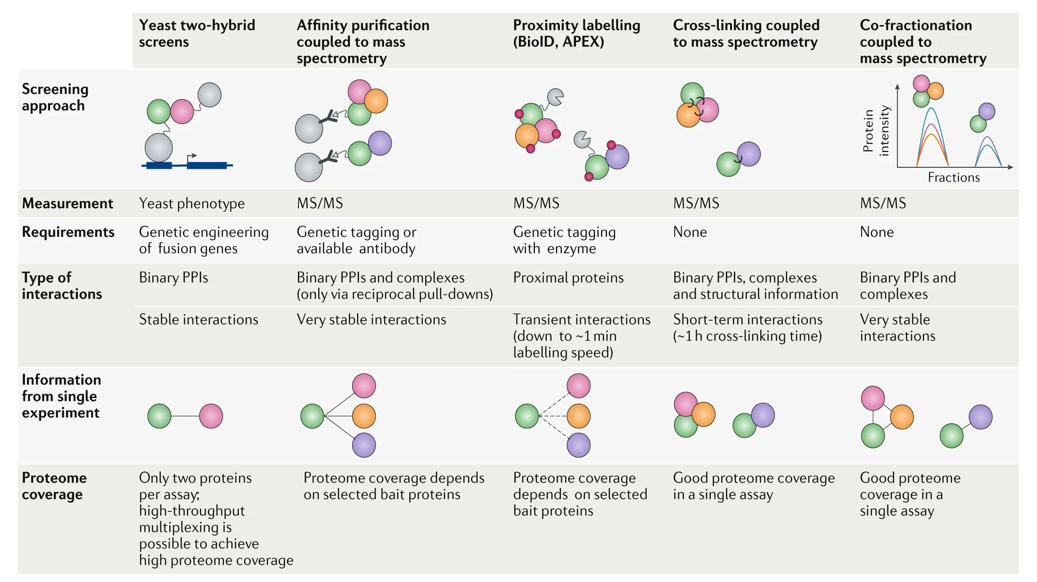
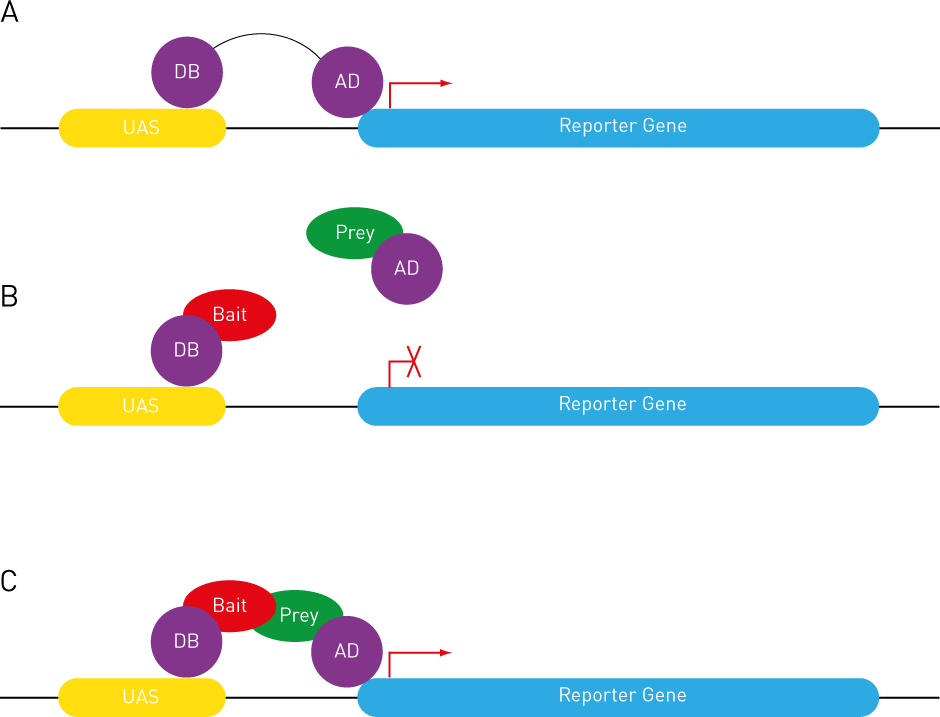
Explain yeast two-hybrid method (what is it, how does it work, limitations)
What?
Molecular biology technique used to detect protein–protein interactions. More specifically, it detects whether two proteins (often called “bait” and “prey”) physically interact within a yeast cell.
How?
The technique exploits the modular nature of transcription factors, which usually have two separable domains:
DNA-binding domain (DBD): binds to specific DNA sequences (e.g. promoter of a reporter gene).
Activation domain (AD): activates transcription when brought near the transcription start site.
In Y2H, the bait protein is fused to the DBD, and the prey protein is fused to the AD. These fusion proteins are then expressed in yeast.
If the bait and prey do not interact, the DBD and AD remain separated, and no transcription occurs.
If the bait and prey interact, they bring the DBD and AD close together. This reconstitutes a functional transcription factor, which activates the transcription of a reporter gene (like HIS3, LacZ, or GFP).
Yeast cells expressing interacting proteins can then be identified by their ability to grow on selective media (e.g. lacking histidine) or to show colour changes (e.g. blue colonies with X-gal if LacZ is used).
Limitations
False positives/negatives are common.
Only detects binary interactions in the yeast nucleus.
May miss interactions requiring specific post-translational modifications or non-yeast-specific conditions.
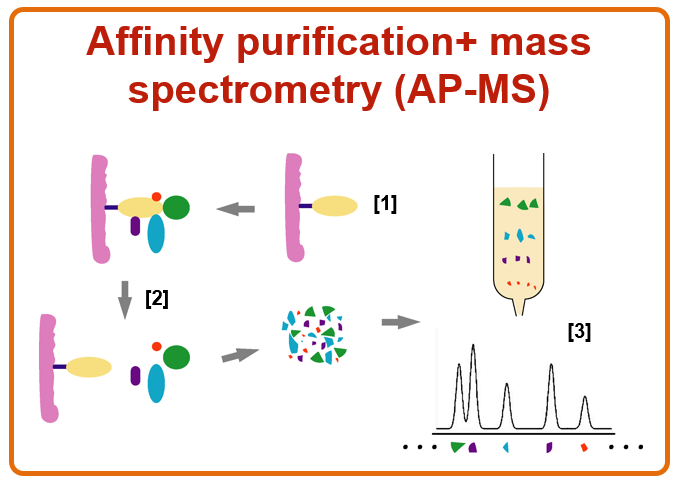
Briefly explain affinity purification coupled to mass spectrometry (AP-MS)
What?
Affinity purification coupled to mass spectrometry (AP-MS) is a powerful technique used to identify and study protein–protein interactions and protein complexes in a more physiological context than methods like yeast two-hybrid.
How?
Affinity purification (AP): A target protein (the "bait") is tagged and expressed in cells. It is then isolated (purified) from a lysate using an antibody or affinity tag. Proteins that are bound to the bait (its interacting partners) are co-purified.
Mass spectrometry (MS): The purified protein mixture is digested (usually with trypsin) into peptides, which are analysed by mass spectrometry to identify the proteins interacting with the target protein.
Main limitation
This technique can miss weak or transient interactions.
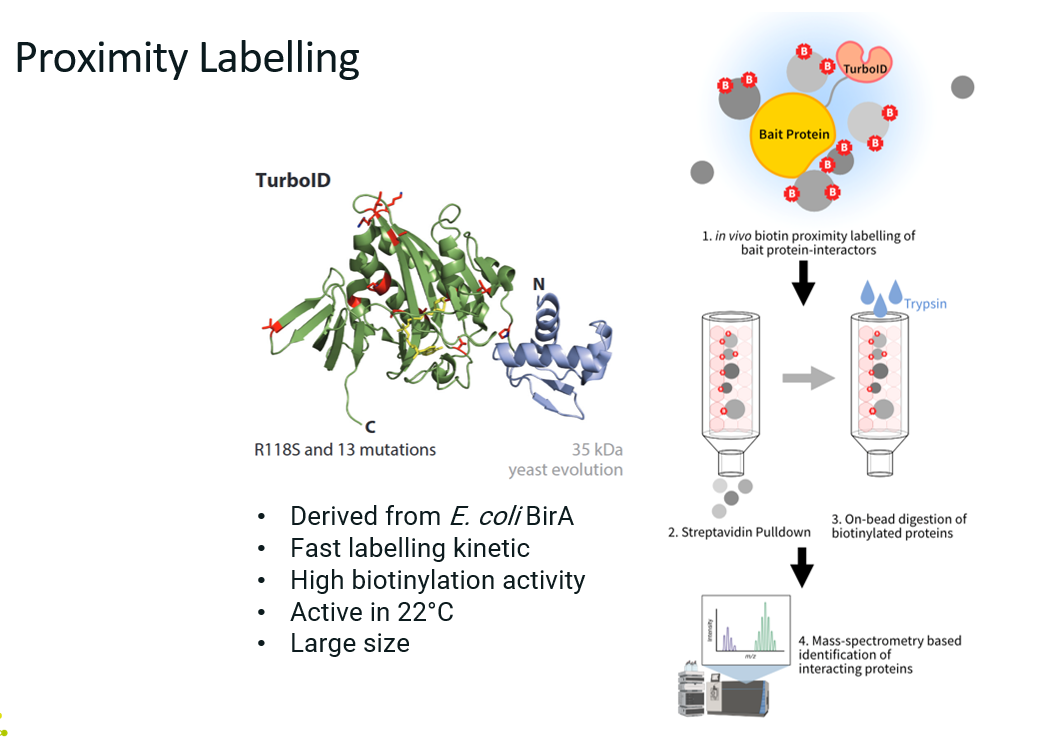
Explain proximity labelling (what is it, how does it work, advantages and limitations).
What?
Proximity labelling is a powerful method for identifying proteins that are spatially close to a protein of interest in living cells, including transient or weak interactors that traditional methods (like AP-MS) might miss.
How?
A protein of interest (the bait) is genetically fused to an enzyme that can chemically ta
g nearby proteins with biotin (e.g. TurboID), not necessarily through direct binding, but via proximity. These labelled proteins can then be easily purified and identified by mass spectrometry.
Clone and express a fusion of bait protein + labelling enzyme in cells.
Add the labelling substrate (e.g. biotin).
The enzyme labels proximal proteins with biotin.
Lyse cells and purify labelled proteins using streptavidin beads.
Elute and identify proteins using mass spectrometry.
Advantages
Captures weak, transient, or indirect interactions.
Reflects spatial proximity, not just physical binding.
Works in vivo, in intact cells or tissues.
Limitations
Background labelling can occur (proper controls are essential).
Requires genetic fusion of bait and enzyme (can affect function).
Don’t know the details on the interactions (was the interaction transient or stable? Did this protein interacted directly or indirectly with the target?).
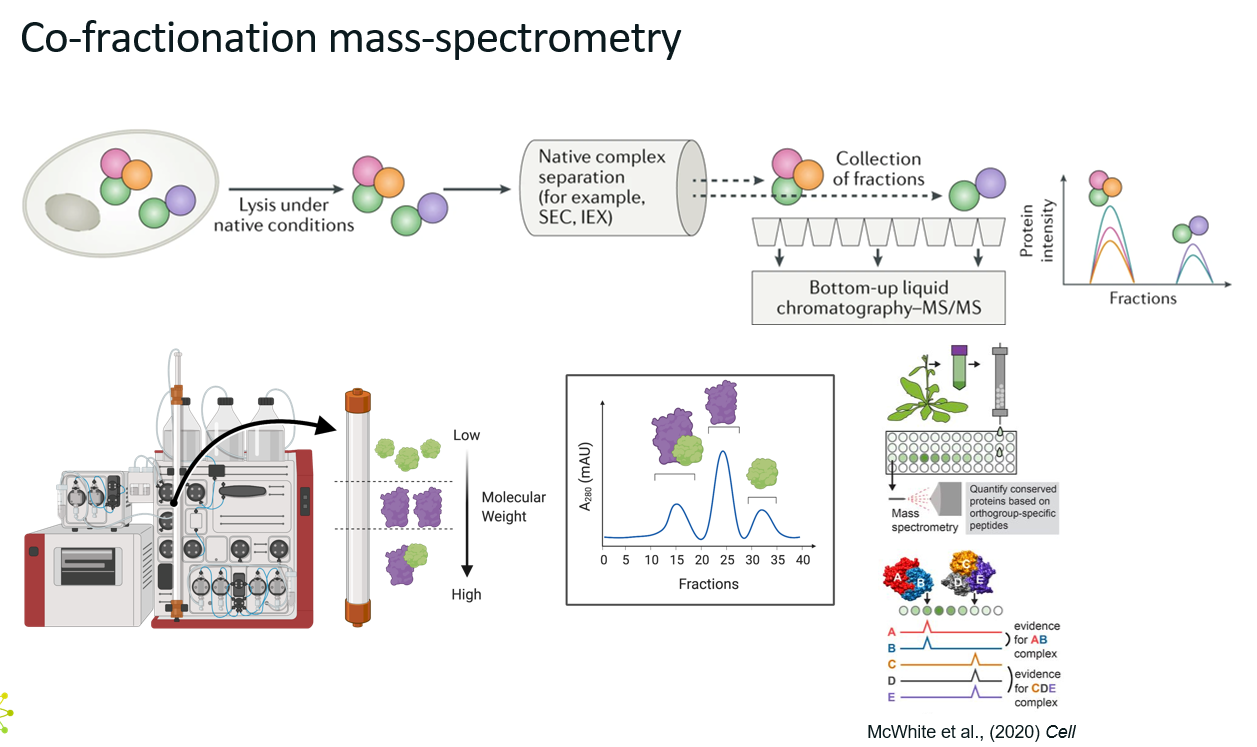
Explain co-fractionation mass spectrometry.
What?
Co-fractionation mass spectrometry (CF-MS) is a method used to identify protein-protein interactions and protein complexes by analysing how proteins co-elute or co-sediment during fractionation.
Unlike methods that rely on tagging or proximity enzymes, CF-MS is label-free, uses native protein mixtures, and leverages the principle that proteins in a complex will behave similarly during fractionation.
How?
Typical workflow
Native cell or tissue lysate is prepared under non-denaturing conditions (so that the protein complexes stay together)
Proteins are separated using a fractionation method such as size-exclusion chromatography (SEC, separate molecules based on size) or ion-exchange chromatography (IEX, separate molecules based on charge).
Each fractions are collected and analysed by LC-MS/MS
Protein abundance profiles across fractions are generated.
Advantages
Suitable for organisms or tissues where tagging is not feasible.
Captures stable and native interactions across conditions.
Limitations
Less sensitive for transient or low-abundance interactions
Large sample and MS workload
When studying transient interactions, the state of the art method is …
proximity labelling.
When studying stable interactions and large complexes, the state of the art method is …
Co-fractionation Mass Spec or Cross linking Mass Spec.
Peptidomics
Peptidomics is the large-scale study of endogenous peptides, short chains of amino acids naturally present in cells or tissues.
Unlike proteomics, which studies full-length proteins and their modifications, peptidomics focuses on naturally occurring peptides.
Examples of important peptides
Oxytocin and vasopressin, which are both neuropeptides (signalling molecules that communicate at the synapses)
Immunopeptides: peptides that are central to the adaptive immune response, because they are what T cells "see" when scanning for infected or abnormal cells.
Challenges in peptidomics
Peptides are very low in abundance.
No trypsin digestion as in proteomics, which means that the peptides have non-tryptic ends and unusual lengths.
Peptidomics workflow
Filter out proteins, only leave behind small molecules, including peptides.
Desalting column to let all the peptides go through and trap all the salts.
Go into mass spec.
Have to have de novo sequencing (no database search)