Lecture 13: Optimising drug properties to ensure good oral bioavailability
1/31
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
32 Terms
what are the requirements of a good drug?
Efficacy o Must achieve the intended therapeutic effect.
Appropriate size and structure to interact with target.
Chemical stability.(over entire
Solubility
Drug needs to be in solution to be absorbed
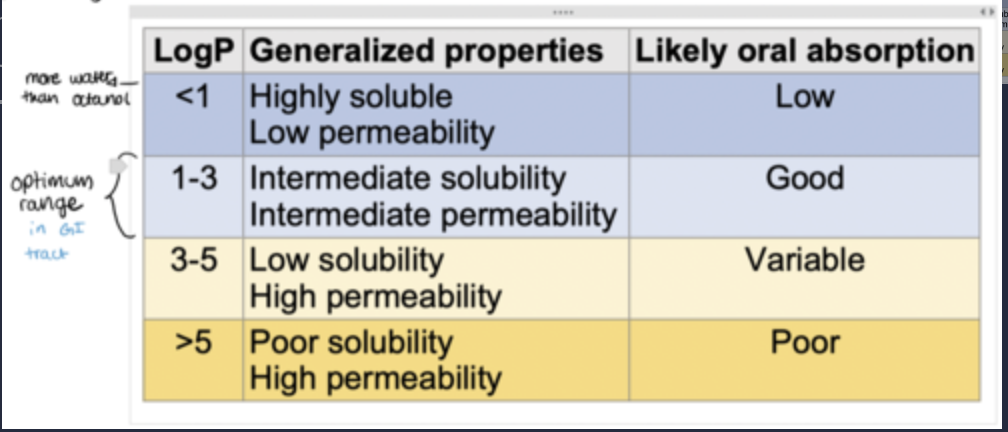
Suitable LogP value (~1-3).
Oral bioavailability.
Appropriate pharmacokinetics.
Favourable safety profile.(not toxic, treating without lots of side effects)
A drug which is highly active in vitro is of no use of it can't be absorbed or if it has poor in vivo performance (e.g. extensive metabolism, toxicity).
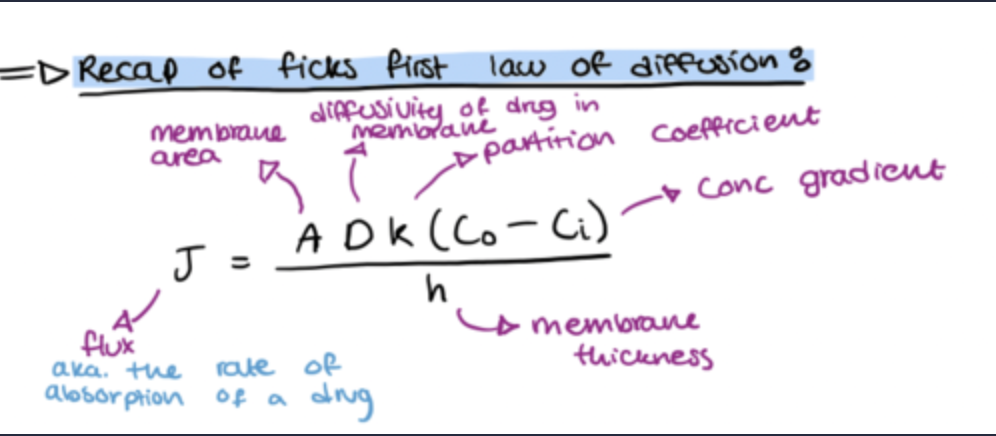
recap of ficks first law of diffusion and how is it used?
Describes drug diffusion across a membrane.
Can also be applied to diffusion rate (flux) across a whole epithelium (considering A and h).
Properties of the drug are K, ΔC and radius (MW).
what is the stokes einstein equation?

We use the partition coefficient (P) of a drug between octanol and water as a substitute for K, the partition coefficient of drug into a biological membrane.
This is a measure of drug lipophilicity and is usually expressed as the log10 value, LogP.
Commonly determined using the "shake flask" method.
Can also be determined by dividing solubility in oil by solubility in water.
More modern methods include use of HPLC and, increasingly, in silico methods.
Values calculated by computer-based methods are known as cLogP.
HPLC compares the mobility of drugs and is much faster however can be less accurate
lipophilicity is the key physicochemical parameter in drug development
what happens if the drug is too hydrophilic?
•If a drug is too hydrophilic, its solubility will be good, but it will be poor at partitioning into the cell membrane.
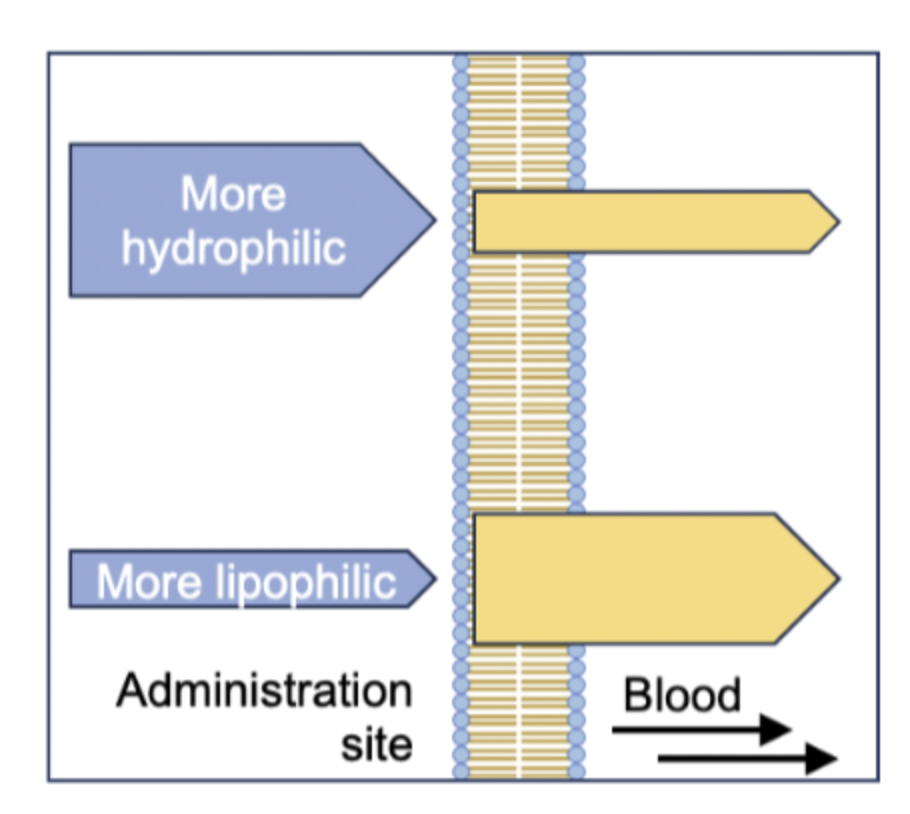
what happens if the drug is too lipophilic?
A drug needs enough lipophilic character to partition into the cell membrane but not be too lipophilic.
If a drug's lipophilicity is too high, it's solubility will be poor, and it will struggle partition back out of the membrane - it may also accumulate in fatty tissues.
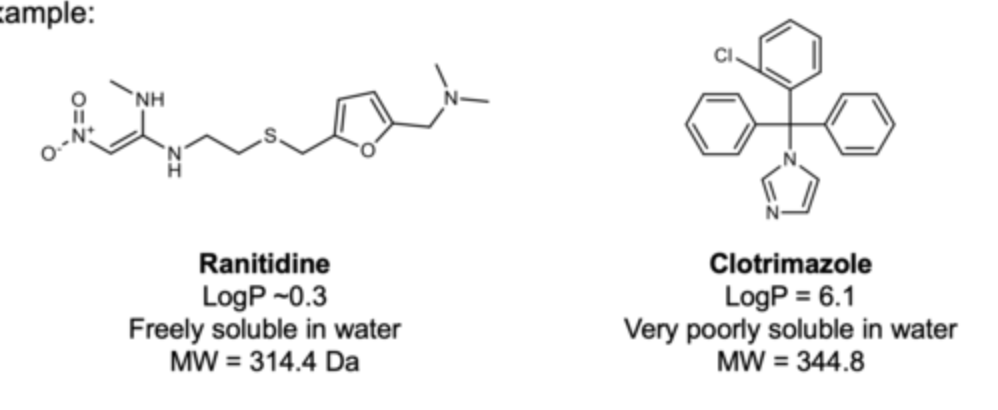
what determines a drugs hydrophilicity or lipophilicity?
The chemical structure of a drug underpins its hydro- or lipophilicity.
For optimal absorption, drug needs a suitable balance of solubility & lipophilicity.
what affects the lipophilicity and LogP of drugs?
A number of aspects of chemical structure affect the solubility and lipophilicity of drugs.
Among these are ionization and hydrogen bonding.
how does pH affect oral drug absorption?
Most small drugs are weak acids or bases - their ionization changes with varying pH according to their pKa.
Ionization affects the lipophilicity of drugs.
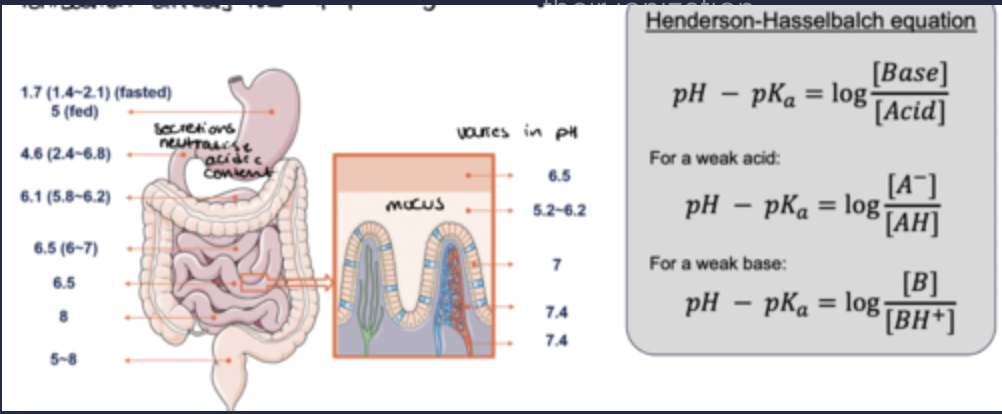
how do ionised drugs affect hydro/lipophilicity?
•Ionized drugs: increased hydrophilicity vs. un-ionized form - reduced membrane permeability.
how do unionised drugs affect hydro/lipophilicity?
Un-ionized drugs: increased lipophilicity vs. ionized form
optimal membrane permeability.
how are strong acids/bases absorbed?
Strong acids (pKa < 3) and strong bases (pKa > 10) are poorly absorbed.
For an acid with pKa of 2 or a base with pKa of 11, only 0.003% of molecules are un-ionized at pH 6.5.
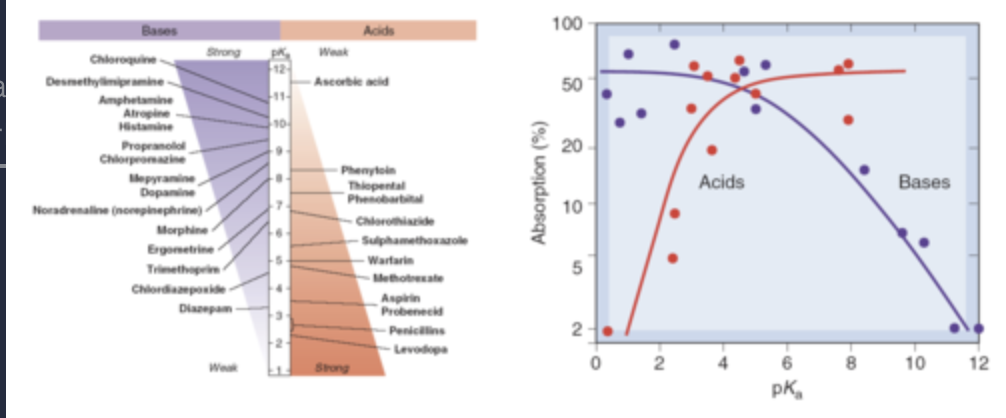
what is the pH partition hypothesis?
AH and B: Un-ionized drug = lipophilic optimal membrane transport
A- and BH+: Ionized drug = hydrophilic reduced membrane transport.
Drug accumulates on the side of the membrane where pH favours ionization. This is the pH partition hypothesis.
what does the pH partition hypothesis not take into account?
A useful concept but doesn't take into account:
Type of epithelium.
Surface area of the absorption site.
Ionized drugs will be absorbed to a small extent.
Active transport of drugs.
Residence time of drug at delivery site.
Charged drugs may form ion pairs with oppositely charged species.
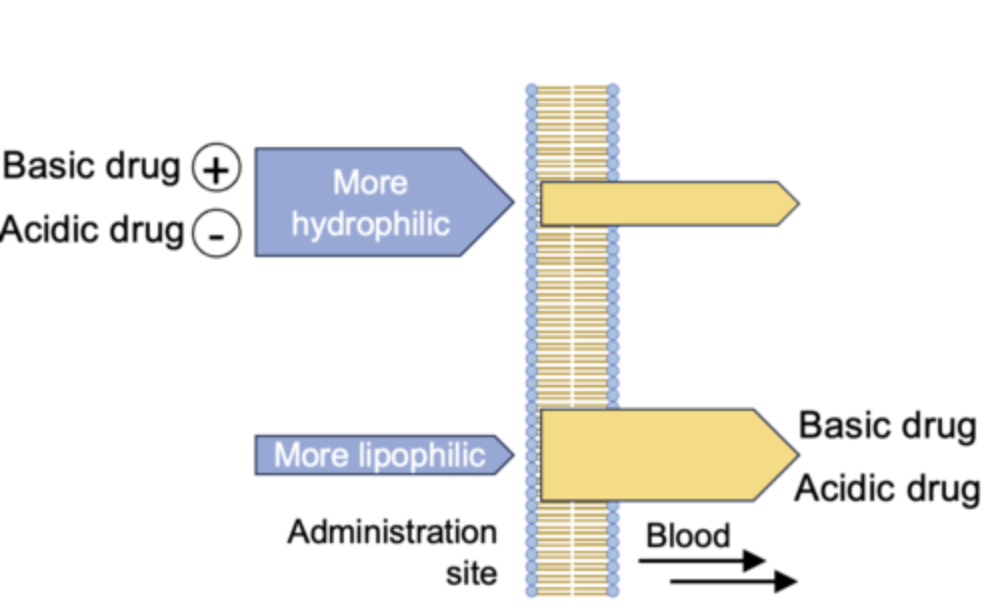
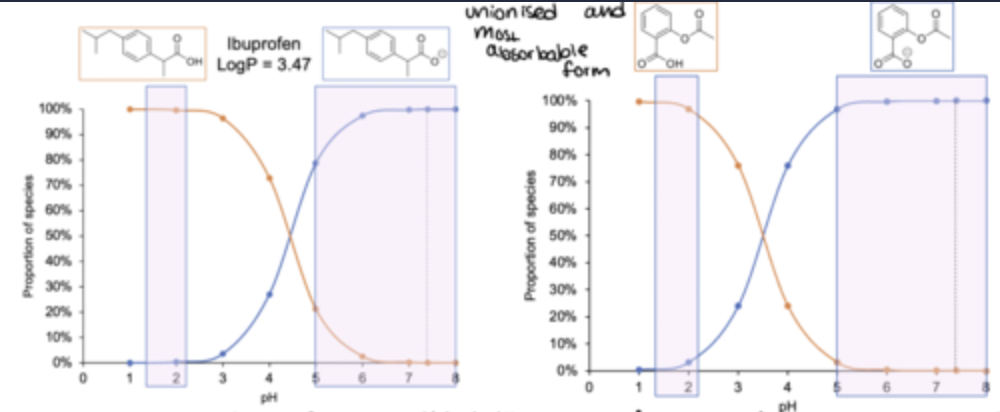
e.g 1: Based on the pH partition hypothesis, will the absorption of ibuprofen be highest in the fasted stomach (pH 2) or small intestine (pH 6.5)?
open notes
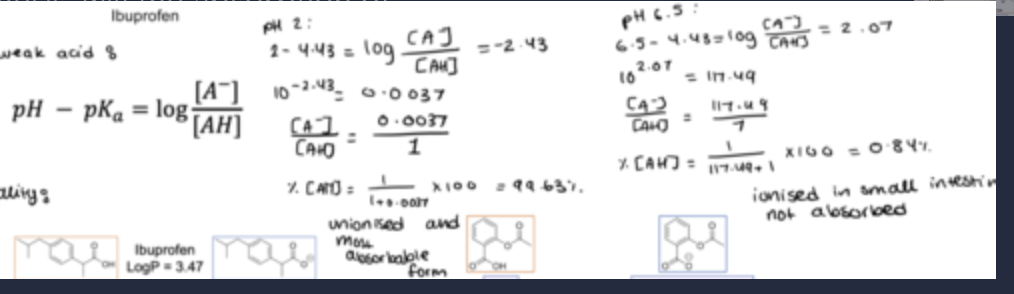
continuation to the example question(graph)
Ibuprofen is un-ionized, therefore more absorbable, in the fasted (empty) stomach - however, its solubility is poor.
Aspirin is un-ionized in the stomach and is more soluble so is absorbed there.
Many drugs are absorbed well in the stomach, but the adaptations of the small intestine mean most absorption occurs there.
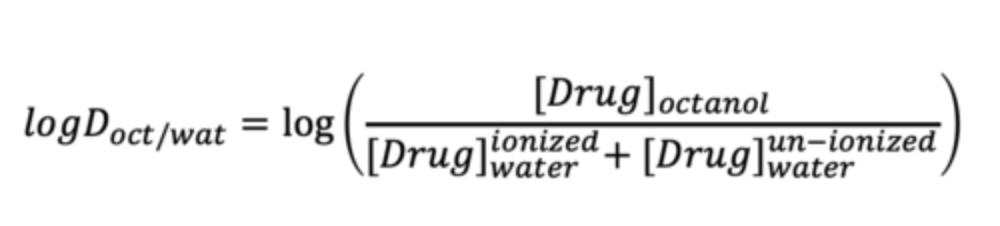
what is an alternative way to measure lipophilicity?
LogP only takes into account the partitioning of neutral, un-ionized species.
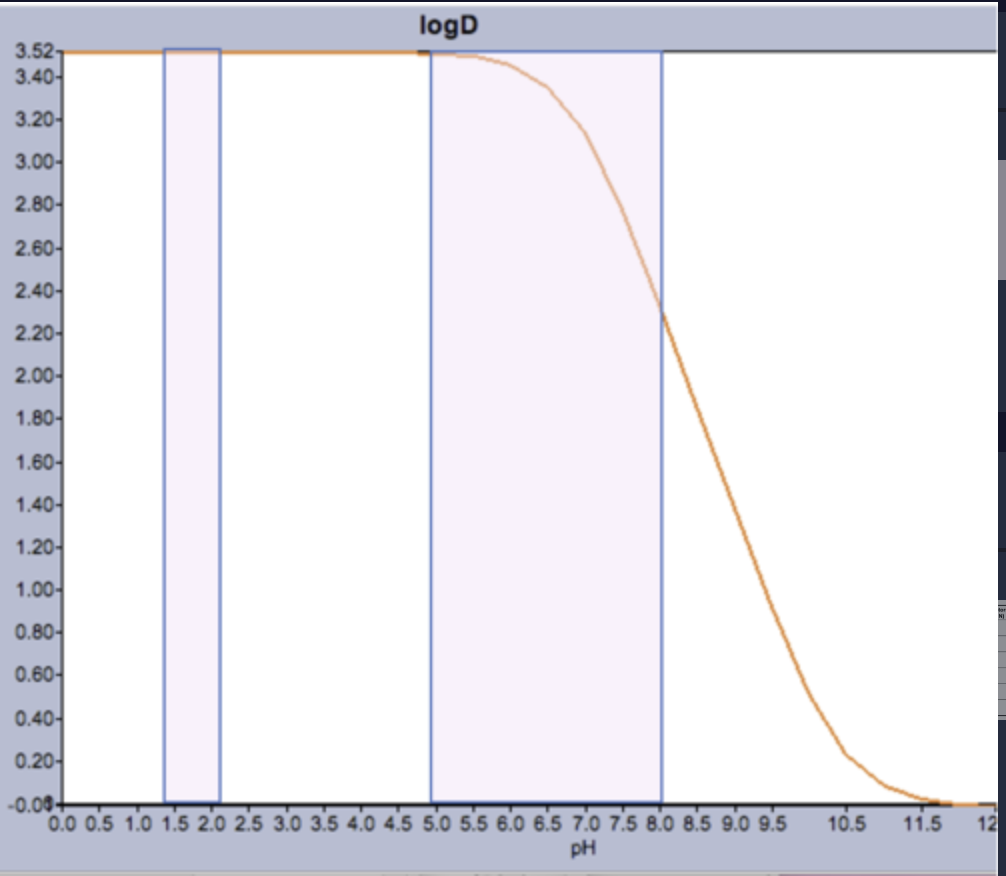
The distribution coefficient, D (expressed as LogD), can give a more realistic measure of lipophilicity by taking charged species into account.
Like LogP, the higher the value of LogD, the more lipophilic the drug is.
LogD can change considerably depending on where in the GI tract a drug is, e.g. the weakly basic drug warfarin (right).
how does hydrogen bonding affect lipophilicity?
it needs to lose water when passing into the membrane
the more H bonding, the more hydrophilic so the poorer the absorption
De-solvation must occur, i.e. hydrogen bonds must be broken, before a drug in solution can enter the lipid plasma membrane.
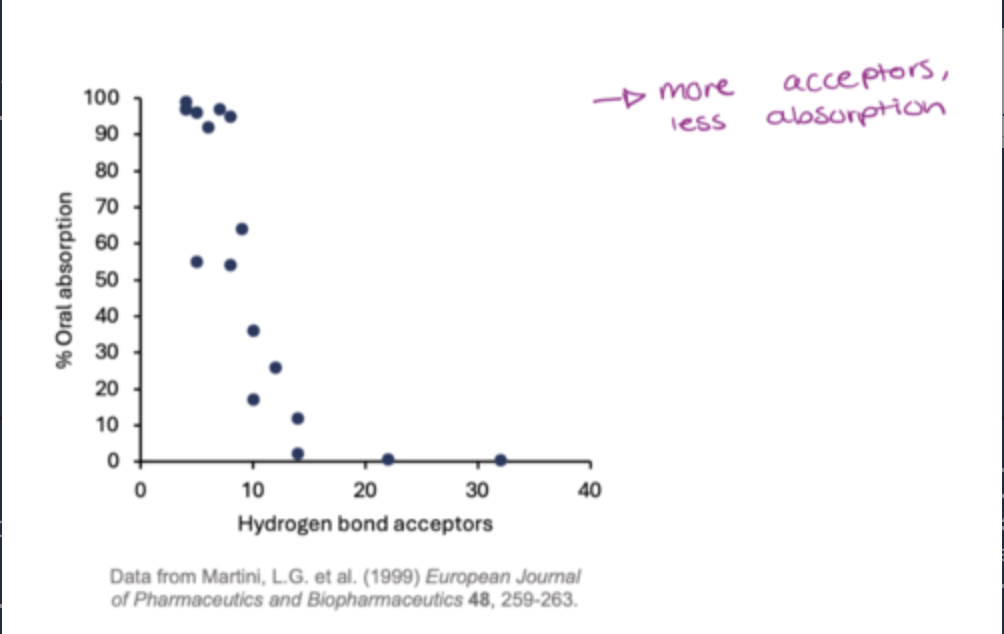
what drug properties does lipinskis rule of five investigate?
Lipophilicity
LogP
Size
MW
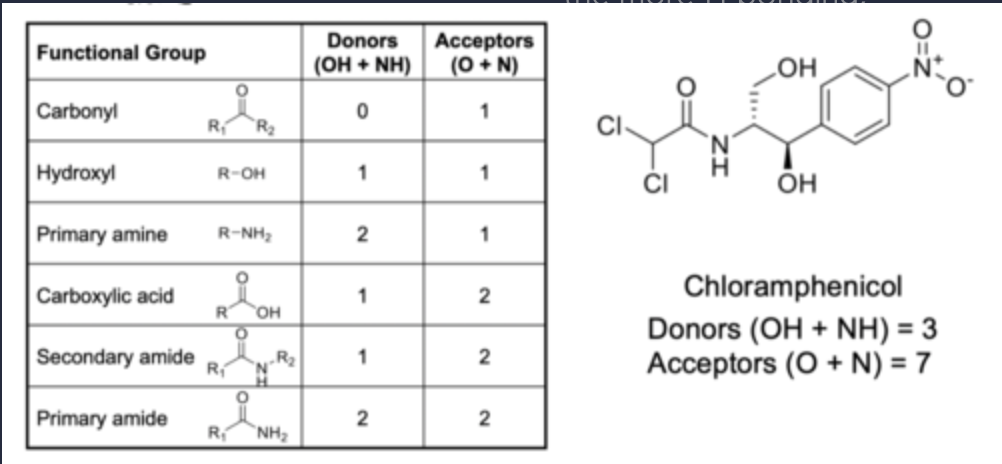
Hydrogen bonding
number of donors and acceptors
The Ro5 predicts that poor oral absorption is likely if two or more of following criteria are broken:
The MW is ≤ 500
The Log P is ≤ 5
The number of H-bond donors (sum of OH + NH) ≤ 5
There number H-bond acceptors (sum of N + O) ≤ 10
all of the following are divisible by 5, hence the rule of 5

what are the exceptions to the rule?
Some drugs break the Ro5 but are still absorbed at acceptable levels.
Most of the drugs which do not comply with the Ro5 are antibiotics
natural compounds or semi-synthetic.
Vitamins, cardiac glycosides and antifungals tend to break the Ro5 too.
Natural products generally adhere to the LogP and H-bond donor requirements.
If oral bioavailability is poor, modified formulations or alternative routes may be used.
examples of the Ro5 being tested:
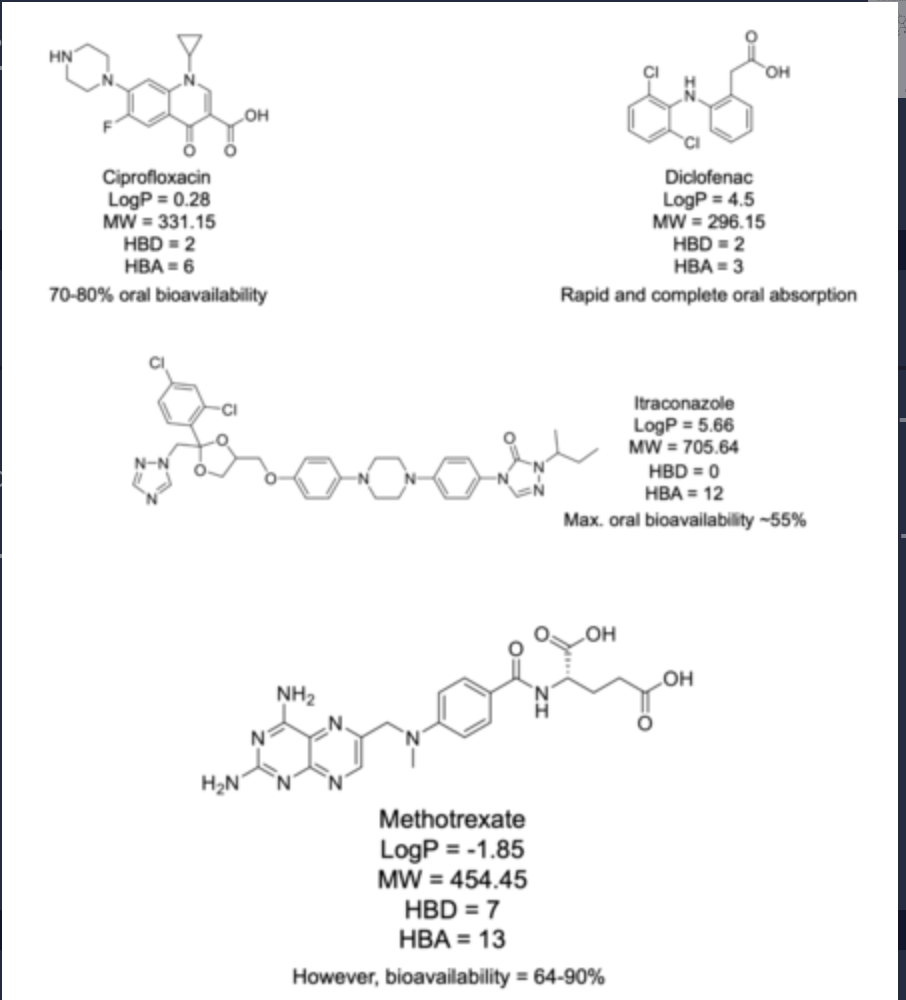
what is lipinskis rule of 5 useful to asses?
Lipinski's Rule of Five is very useful for assessing the likelihood of a drug being well absorbed following oral delivery.
However, there are alternative approaches and "rules" which can be used in combination with (or instead of) the Ro5. For example, Veber's Rules
what are vebers rules?
Number of rotatable bonds ≤ 10
Total polar surface area (TPSA) ≤ 140 Å2 or total H-bond count ≤ 12.
the lower the better for both
the bigger the molecule, the more the rotatable bonds and the higher polar SA.
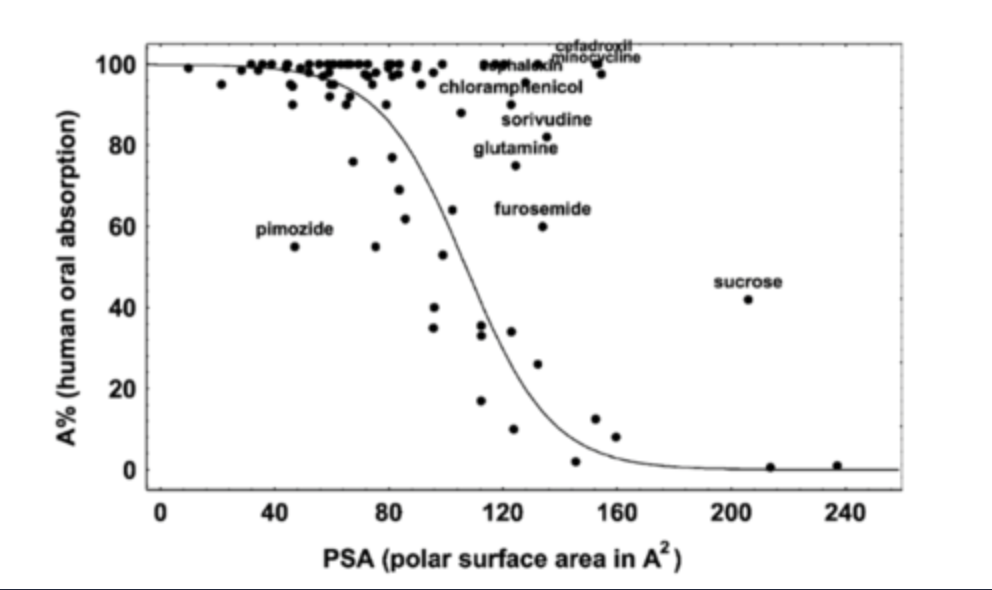
optimising drug properties for oral absorption: what if a drug which has excellent activity in vitro has poor bioavailability?
It may be possible to make chemical modifications to alter polarity or LogP assuming this doesn't affect binding to its target.
This can include:
Incorporation of ionizable groups to increase solubility.
Changing the pKa functional groups to increase lipophilicity.
Reducing the number of hydrogen bond donors or acceptors.(changing functional groups)
Pro-drug strategies.
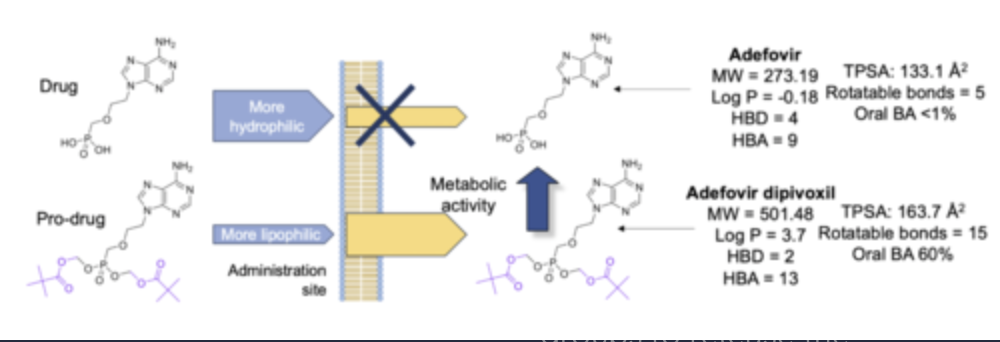
If a drug is too hydrophilic, oral absorption will be limited due to limited permeability. how can you improve its lipophilicity and membrane permeability?
drugs can be chemically modified with lipophilic groups (or groups which target transporters).
Following absorption, metabolic processes within cells cleave the lipophilic groups to release the active drug.
This is known as the pro-drug approach, and many common medicines contain pro-drugs rather than the true API.
summary:
read
