GI 2.2 Final
1/49
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
50 Terms
what is the process of heme anabolism (production)?
glucose → G-6P → 3P glycerate → serine → glycine → heme
how is glycine processed into heme?
glycine → ALA → porphobilinogen → hydroxymethylbilane → uroporphyrinogen III → coproporphyrinogen III → protoporphyrinogen III → protoporphyrin → heme
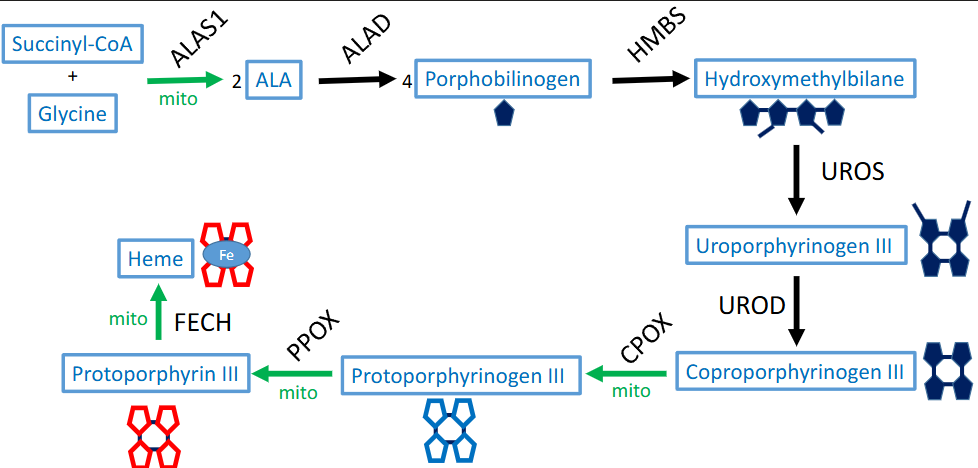
what is the rate limiting step of heme anabolism? what regulates it? products?
glycine (+succinyl coa) → ALA
first step, translationally regulated
via ALAS1 + cofactor (B6)
also produces CoA-SH, CO2
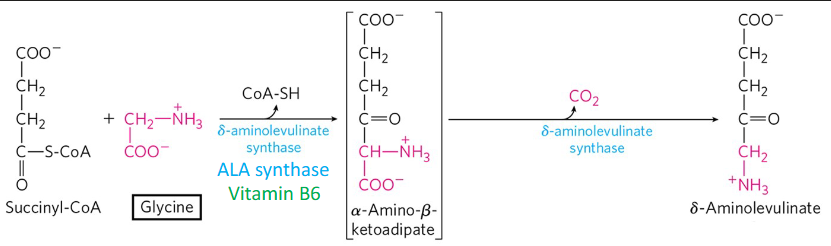
how does iron effect RDS of heme anabolism? low iron?
Fe translate AL to allow heme production
low Fe =mRNA initiation site blocked + NO ALA translated (BAD)
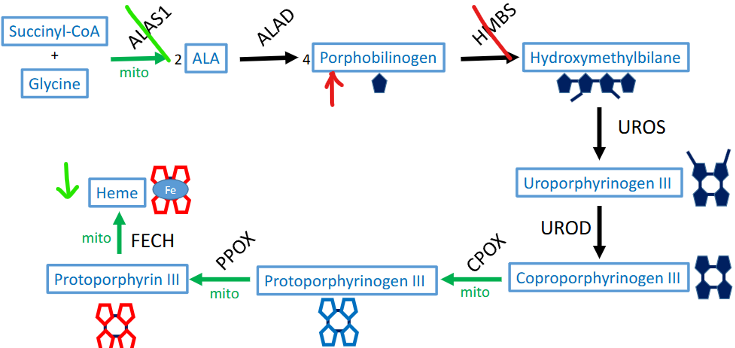
what causes acute intermittent porphyria (AIP)? symptoms? treatments?
def. HMBS (hydroxymethylbilane synthetase) via high liver demand (infection, fast, alcohol, steroid)
symptoms: abdominal pain, psych. issues, peripheral neuropathy, precipitated, pink urine (5Ps)
ALA + porphobilinogen inc. in urine → pink
hemin (panhematin) → block ALA synthase (interfere w/ porphyrins + heme synth.)
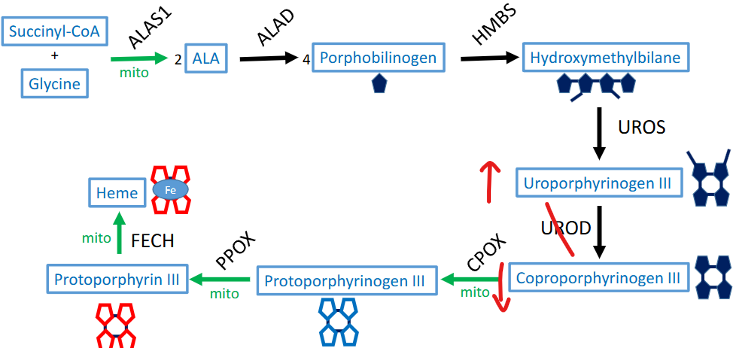
what causes porphyria cutanea tarda (PCT)? symptoms?
UROD (uroporphyrinogen decarboxylase) def. or inhibitors
uroporphyrinogen III accumulate + coproporphyrinogen III def.
symptoms: photosensitivity, blistered skin, red urine (via uroporphyrin)
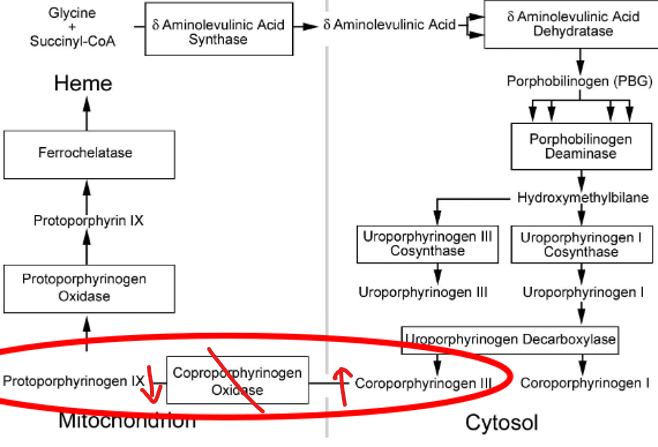
what causes variegate porphyria (VP)? symptoms?
acute hepatic
mut. PPOX (protoporphyrinogen) → PPIX excess in urine
dx: inc. plasma porphyrins
symptoms: photosensitive + blister, neurovisceral attack, inc. ALA synthase
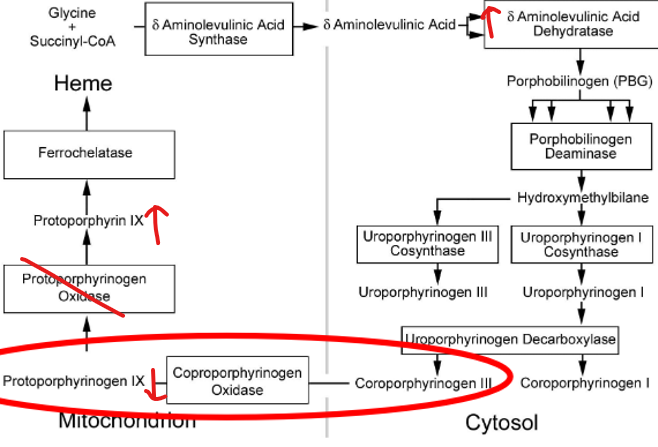
what causes hereditary coproporphyria (HCP)? symptoms?
acute hepatic
mut. CPOX (coproporphyrinogen) → CPIII excess in urine
dx: fecal coproporphyrins
symptoms: same as VP (less severe cutaneous)
what type of medications do VP and CP precipitate with?
CYP450 inducers
phenytoin + rifampin
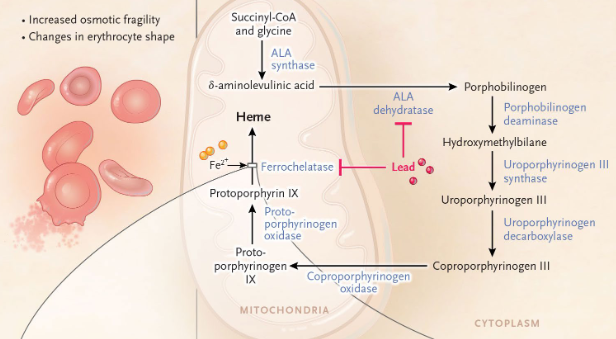
what does lead poisoning cause? what does it do to Ca? symptoms? prevention?
inhibition of enzymes w/ Cys (ie ALA DH + ferrochelatase) → elevated ALA + Fe
replaces Ca in proteins → block NT firing→ dec. neuronal connection
symptoms: cognitive issues, fatigue, blueness, bone/muscle dev. issue, hydrochromic microcytic anemia (w/ basophilic stippling)
lead pipe w/ HPO4 = lead solidify → not in drinking water
what is the process of heme catabolism?
MO engulf heme → biliverdin (via heme oxygenase) → bilirubin (via biliverdin reductase) → stercobilin + urobilin
what is the difference between direct and indirect bilirubin?
direct → conjugated
via UGT-1 (UDP GT) adding glucuronic acid = inc. solubility
also blue light (biliverdin photosensitive)
indirect → unconjugated
insoluble (more toxic), bound to albumin (to liver)
what are the OA transporting peptides holding bilirubin in hepatocytes?
SLCO1B1 + 3
what are phases of a bruise?
1 → pink + red (via hemoglobin)
2-6 → blue + purple (via hemoglobin)
7 → pale green (via biliverdin)
8-14 → yellow + brown (via bilirubin)
what are symptoms of hyperbilirubinemia? types?
jaundice + scleral icterus
types:
unconjugated → UGT-1A def.
conjugated → overactive UGT-1A
what is crigler-najjar T1? T2?
unconjugated hyperbilirubinemia
T1 → absent UGT-1A
unconjugated bilirubin → BBB → encephalopathy → kernicterus (fetus, fatal)
T2 → reduced UGT-1A
treat w/ phenobarbital (makes more UGT-1A)
what is gilbert syndrome? neonatal jaundice?
unconjugated hyperbilirubinemia
gilbert → mild dec. UGT-1A (benign)
neonatal → 2-5 days post birth via low UGT-1A
light therapy conjugates bilirubin (H2O soluble photo isomers) = dec. bilirubin + prevents kernicterus
what causes dubin-johnson syndrome?
conjugated hyperbilirubinemia
ABCC2 mut. → dec. MRP2 transporter → prevent conjugated bilirubin sec. (hepatocyte → bile duct)
or pigment gallstone (block bile duct)
black liver
MOST BENIGN
what causes rotor syndrome? symptoms?
conjugated hyperbilirubinemia
SLCO1B1 + 3 mut. → dec. OA transport polypeptide act.
= less conjugated bilirubin in hepatocyte + more in circ. → high mixed
red urine via coproporphyrin I
what is the first barrier of the immuno system?
simple squamous → stomach, intestine, biliary tract
pseudostratified columnar → trachea, bronchi, bronchioles
NK strat. squam. → nasal sinus, tonsil, pharynx, larynx, oral cavity, esophagus, rectum
what are the cells within the mucosa?
goblet cells → sec. mucus
paneth cells → AMPs (defensin), lysozyme, lactoferrin, phospholipase A2
brunner glands → sec. HCO3
what are the paneth cells defensins composed of?
a → neutrophils, NK cells, paneth cells located in crypts of SI
B → epithelial cells of skin + mucosa, immune cells
expression upregulated by infection, cytokine, IBD
what are MALTs? MALT lymphoma? cause? symptoms?
adaptive immune response against pathogen @ mucosal sites
lymphoma → chronic antigenic stim. of marginal B cells
cause: chronic infection (H. pylori) or autoimmune (sjorgren)
symptom: upper gastric pain, weight loss, fatigue, erythematous mass on funds (endoscopy)
what are GALTs?
peyers patch (ileum)
mesenteric LN
large + small lymphoid aggregates (L: appendix, colon) (S: esophagus)
lymphoid cells in LP
what type of cells do GALTs consist of? function?
B + T cell, MO, DC, mast cells
fxn: sample luminal antigens via
M cells: peyers patch, transcytosis → lymphoid → FDC/DC uptake
DC: LP, extend processes
sIgA: w/in binding antigen, transcytosis thru M cell → GALT → FDC/DC uptake
IgG: binds antigen + inwards transport via FcRn
what are the innate immune factors?
gut microbiota
AMP → defensins, lysozyme, lactoferrin, phospholipase A2
PRR (TLR + NLR) → limit unnecessary inflammation
tolerogenic MO → T reg attenuate inflammation via IG10 + TF
IL10 def. → diffuse inflammation via no attenuation
ILC → interleukins
what are the types of ILCs?
ILC1 → IFNy + TFNa → MO act. + elim. of IC infection (bacteria/virus), epithelial damage
ILC2 → IL4, IL5, IL9, IL13 → eosinophilic act. + mucus prod., helminth infection
ILC3 → IL17 + IL22 → defensin/AMP → enhance mucosa fxn + immune response, EC pathogen
what is the humeral adaptive immune response?
IgA + IgM bind pIgR
IgG binds FcRn
homing of IgA producing B cells: TSLP → RALDH → retinoic acid → CCR9 → CCL25 → a4B7 → MAdCAM
what are the cellular (IEL) adaptive immune factors?
natural IEL → CD4-CD8-TCRaB+ T cells, CD4-CD8-TCRyo+ T cells
induced IEL → CD8+ T cells, CD4+ T cells, TCRaB+ T cells
what are the cellular (T cell) adaptive immune factors?
Treg → TGF-B + IL10 (attenuate)
Th2 → IL4, IL5, IL9, IL13 (eosinophil act. + mucus for helminth)
Th17 → IL17, IL22 (defensin/AMP + EC pathogen response)
what are the lymphatic adaptive immune factors?
a4B7 (LPAM-1) → MAdCAM-1 (mucosal HEV endothelium)
CD62L (L selectin) → GlyCAM-1 + CD34 (all HEV endothelium, weak)
LFA-1 → ICAM-1 (all HEV endothelium, strong)
CCR7 → CCL19, CCL21 (all HEV endothelium, chemotaxis)
what causes celiac disease? symptoms? what does it causes risk of?
HLA-DQ2/8 mut. → tissue-transglutaminase (TTG) antibody response against gluten + villous atrophy
symptom: growth delay, diarrhea, belly pain, irritability, muscle pain, anemia, dermatitis, herpetiformis
@ risk for vit. D, B, Fe, Ca def.
what causes whipple? symptoms? histological examination?
HLA-B27 haplotype w/ gram (+) tropheryma whipplei infection → GI malabsorption
symptoms: weight loss, abdominal pain, diarrhea, arthritis (WADA) → can involve CV, CNS, joint
histological → foamy MO in intestine
what is IBD? symptoms?
failure of oral/gut mucosal tolerance, ass. w/ gut dysbiosis + genetic predisposition
symptoms → intermittent diarrhea, abdominal pain, rectal bleed/bloody stool, weight loss, malnutrition
what is the difference between UC and CD?
UC → cont. inflammation of colon to rectum (some cecum)
CD → scattered transmural inflammation on term ileum + colon (cobblestone)
fissures, fistulas, obstruction, abscess, perforation, sinus, stricture (FOAPS)
skip lesion, ileum + colon, perianal, spare rectum (SKIPS)
dense lymphocyte collection, defect in B defensin production
what causes type 0 GSD? symptoms
def. glycogen synthase
symptoms: severe fasting hypoglycemia (glycogen synthase is RLS of glycogenolysis)
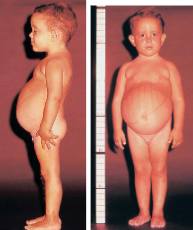
what causes type 1 GSD (von Gierke)? symptoms?
inc. G-6P in liver cells (muscles spared) → hypoglycemia (early death, seizures), lactic acidosis, liver enzymes normal
type IA (G-6Pase def.)
hepato/renomegaly, doll like face
need NGT tube for glucose
type IB (G-6P transporter def.)
diarrhea, bruising, neutropenia
what causes type II GSD (pompe)? symptoms?
def. a-glucosidase (GAA) or acid maltase → dec. glycogen breakdown in lysosome (muscle)
symptoms: cardiomegaly, floppy infant syndrome (hypotonia, low CK), macroglossia, resp. issues
high CPK, AST, LDH, glycogen filled vacuoles
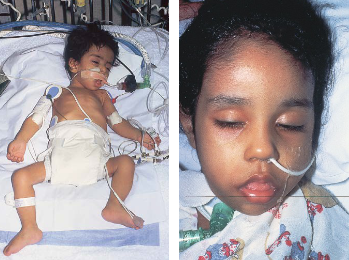
what causes type III GSD (cori/forbes)? symptoms? how does it differ from type I GSD (von Gierke)?
def. a-1,6-glucosidase (debrancher) (liver + some muscle)
mild von Gierke BUT: less severe hypoglycemia, NO lactic acidosis
weak bones, skeletal/cardiac muscle weakness, fasting ketosis (ketones in urine), inc. AST/ALT
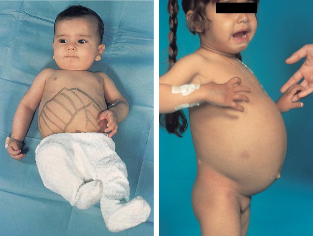
what causes type IV GSD (andersen)? symptoms?
def. glycosyl 4,6 transferase branching enzyme (GBE1)
symptoms: neuro impact, elevated bilirubin, prolonged PT/INR
what causes type V GSD (McArdle)? symptoms?
def. muscle glycogen phosphorylase (myophosphorylase) → removes glucose form outer branch (muscle)
symptoms: burgundy urine, exercise intolerance (cramps), “2nd wind” after VD
inc. CK
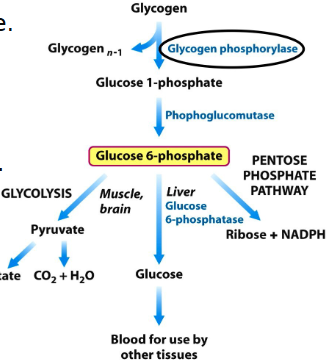
what causes type VI GSD (hers)? symptoms?
mut. liver glycogen phosphorylase
symptoms: hepatomegaly, hypoglycemia, fasting hyperlipidemia+ ketosis
better after eating
what is the exocrine and endocrine function of the pancreas?
exocrine → pancreatic juice w/ enzymes digesting carbs (amylase), proteins (trypsin), fats (lipase)
endocrine → islet cell
a → glucagon
B → insulin, amylin
D → somatostatin
what causes acute pancreatitis? how does necrosis occur?
trypsinogen + chymotrypsin early act. w/in pancreas → autodigestion inflammation + hemorrhage (severe= necrosis)
damage = lipase release from acinar cell → saponification necrosis
what is the difference between interstitial/edematous pancreatitis and acute hemorrhagic pancreatitis?
interstitial/edematous → mild inflammation w/ edema + wide interstitial space (85%)
no necrosis, not Tx, spontaneous heal
acute hemorrhagic → severe, prominent enzyme mediated tissue destruction (15%)
what are the etiologies of acute pancreatitis? diagnosis? imaging?
idiopathic, gallstone, EtOH, trauma, steroids, mumps, autoimmune, scorpion bite, hypertriglyceridemia/hypercalcemia, ECRP, drugs (valproic acid azathioprine + diuretics)
Dx: severe epigastric pain radiating to back, nausea/vomit, inc. amylase/lipase/trypsin
US w/ enlarged hypoechoic pancreas or CT w/ contrast showing pancreatic necrosis, inflammation, retroperitoneal fluid
what is chronic pancreatitis? labs? US? etiologies?
persistent pancreas inflammation → impair endocrine fxn
labs: inc. serum lipase, amylase (sometimes bilirubin + ALP if bile duct compress)
US → calcification, ductal dilation, pancreatic enlargement, peripancreatic fluid accumulation
etiologies → gallstone, alcohol, hypertriglyceridemia/hypercalcemia, tumors blocking cyst, cystic fibrosis
pancreatic cancer symptoms? risk factors?
courvoisier sign (distended GB), trousseau sign, sister mary-joseph node
risk: hereditary pancreatitis, BRCA2, peutz-jeghers, ataxia telangiectasia, ABO blood, chronic pancreatitis, smoking, obesity, NSAIDs, H. pylori, infection HepB
pancreatic cancer symptoms? imaging?
symptom: epigastric pain radiating to back, weight loss, obstructive jaundice, steatorrhea, new onset diabetes (asymptotic until invasion)
imaging: endoscopic US, percutaneous pancreatic biopsy (FNA), CT, ERCP, MRCP, MRI
cholelithiasis? cholecystitis? choledocholithiasis? cholangitis? cancer?
cholelithiasis (gallbladder stones) → light stool/dark urine
cholecystitis (inflammation) → + murphy sign
US, MRCP (dilation), ERCP, Tx: cholecystectomy
choledocholithiasis (bile duct stone) → cut. jaundice + scleral icterus, hyperbilirubinemia
ERCP
cholangitis (bile system inflammation) → charcot triad (RUQ, fever, jaundice)
cancer → calcification/fibrosis (porcelain GB)