PSC 4215 - Exam 2 - Combined set - General principles of drug disposition, Movement across membranes, Review of absorption, Distribution of drugs, PLP plasma proteins
1/204
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
205 Terms
Which substances are absorbed in the stomach, and why?
Weak acids and alcohol remain unionized in the stomach’s low pH (acidic).
Unionized molecules are lipophilic so it can cross the lipophilic cell membrane by passive diffusion and absorb into tissues
Which drugs act rapidly and why? Provide an example.
Lipophilic drugs bc it can cross the lipophilic cell membrane quickly
Anesthesia
Describe oral drug journey from ingestion to excretion.
Stomach = Drug dissolution
Intestines = Drug absorption
Liver = 1st pass metabolism (reduces drug concentration)
Systemic circulation/blood = Distributes drug to target tissues
Target site = Drug produces effect
Excretion = Urine (via kidney filtration), feces (via liver metabolism)
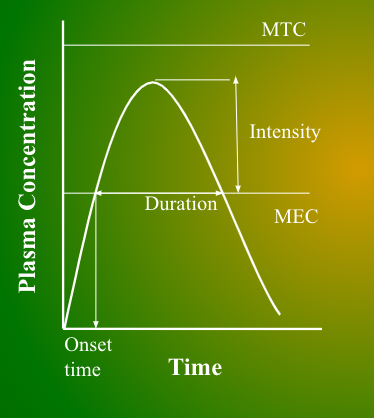
Define onset, duration, intensity of pharmaceutical agents
Onset = Start time
Duration = Length
Intensity = Strength
What determines the magnitude of a drug?
Dose
The higher the concentration at the target site, the higher the intensity
(Amount of drug taken, ex; 500mg.)
Does higher dose lead to faster onset? Why/why not?
No, onset depends on absorption speed
Pharmacokinetics (PK) vs. Pharmacodynamics (PD)
PK = What the body does to the drug (ADME)
PD = What the drug does to the body
ADME
Absorption, distribution, metabolism, excretion
ADME is the study of what pharmacologic process?
PK
Effect of a drug on target cells is called?
PD
What is a xenobiotic? What kind of substance can act like it?
Foreign substance
Endogenous substances (ex: Taking thyroxine externally due to thyroid gland removal.)
How are doses established in clinical trials? What 3 factors does testing include.
Trial & error in phase 2 of FDA approval process
Testing includes dose, interval, route
T/F: Drug effect is proportional to concentration at site of administration.
False. Drug effect is proportional to concentration at the site of action (where the drug works) not administration (where the drug enters)
What are the two sites where concentration gradients are measured in PK?
Site of administration and site of action
T/F: Drug destroyed post-absorption still has high effect.
False. If a drug is absorbed, then destroyed by first pass metabolism before reaching target site, it will have a low effect.
Why do many drugs cause side effects?
They reach non target sites during distribution
What do acidic drugs bind to?
Albumin (the main plasma protein)
What do basic drugs bind to more?
α1-acid glycoprotein
Define Free drug. How is it eliminated?
Active drug molecules that can cross cell membranes and bind to receptors.
Eliminated thru kidney filtration (excreted as urine) or liver metabolism (taken up by enzymes, excreted as bile or feces)
Protein bound drug
Inactive complex that acts like a reservoir bc it slowly releases free drugs
Increases drug presence in the blood
When plasma proteins bind to drugs, what is the effect on duration and elimination?
Increases duration bc the complex stays longer in the body
Reduces elimination bc complex is too large for elimination or absorption
Why exclude albumin-bound drug from effect curves?
Complex is inactive
What are 4 fates of a free drug in plasma?
Effect
Sequestered (absorbed)
Metabolized
Eliminated
4 main drug elimination routes?
Urine, feces, sweat, exhaled air
What does concentration vs time curve show?
X = Time
Y = Concentration
Up/down (curve) = Intensity
Up = Rising drug levels (Due to drug absorption/injection)
Down = Drug elimination (Due to metabolism/excretion)
Left/right = Half life (The amount of time the drug in the body reduces of half)
Left = Narrow width of the curve due to steep slope indicates short half life (good for short exposure)
Right = Wide width of the curve due to gentle slope indicates long half life (good for long exposure)
Left/right = Onset
Left curve shift = Faster onset
Right curve shift = Slower onset
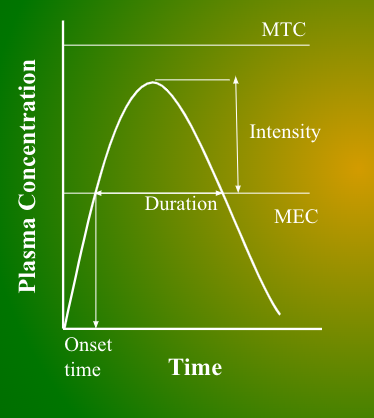
6 Methods of collecting drug concentration data
Urine, feces, exhaled air, blood, CSF, biopsy
Blood vs. plasma vs. serum — difference?
Blood = Whole blood cells (RBC, WBC, Platelets) + Plasma (liquid portion)
Plasma = Liquid portion only + Clotting factors (No blood cells)
Serum = Clotted plasma
T/F: Drugs in blood are in the water portion.
True, unbound drugs (free drugs) remain in the plasma
T/F: Total body drug amount can be measured directly.
False, only estimates are possible by measuring plasma
Positive vs. negative slope in concentration-time curve?
Positive = Absorption > Elimination
Negative = Elimination > Absorption
What does a plateau in the concentration-time curve mean?
Absorption = Elimination
Why does plasma concentration reflect tissue concentration?
They are in equilibrium
Free drugs move freely between plasma and tissues, so plasma concentration mirrors tissue concentration
MEC
Minumum effective concentration
Below this, there is no therapeutic effect
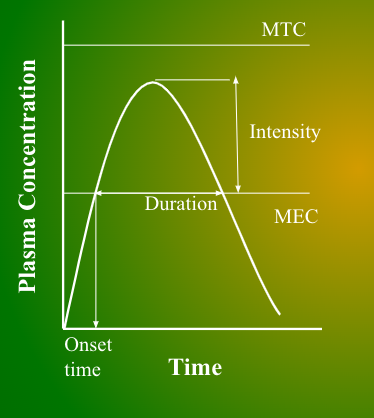
MTC
Minimum toxic concentration
Lowest level for toxicity
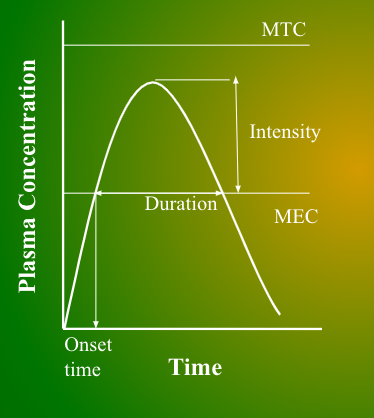
How to find drug duration on a conc-time graph?
The start and end time of the drug concentration that reached MEC
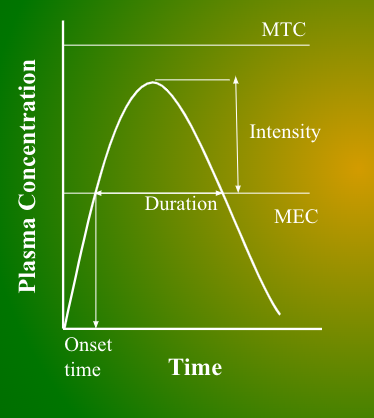
How to find drug onset on a conc-time graph?
The time that drug concentration reaches MEC
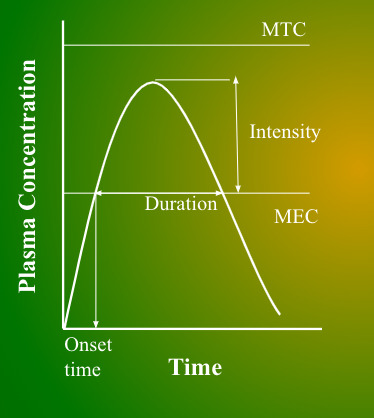
T/F: MEC always gives desired response.
False. MEC marks the start of the therapeutic response not the max
Response intensity is proportional to what?
Plasma concentration
AUC
Area under the curve
Total amount of the drug that is absorbed systemically
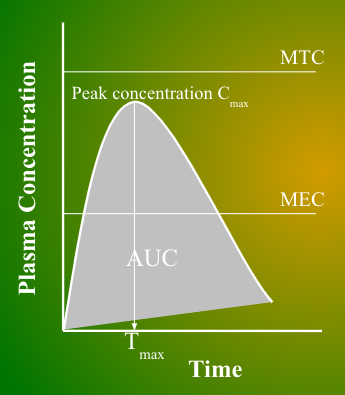
On a conc-time curve, does the curve shift right/left with IM injection?
It shifts the curve left which indicates faster onset
Tmax vs. Cmax
Tmax = The time it takes to reach max (peak) concentration in the plasma
Cmax = Max (peak) concentration in the plasma
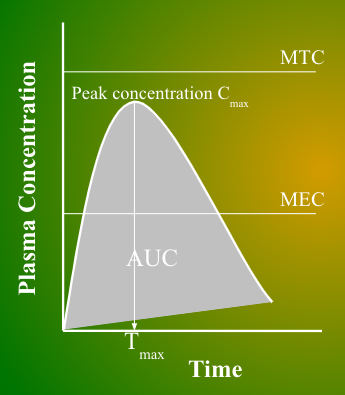
Effect of IV bolus on conc-time curve?
It starts very high with a steep negative slope
Effect of infusion on conc-time curve?
Rises slowly then plateau
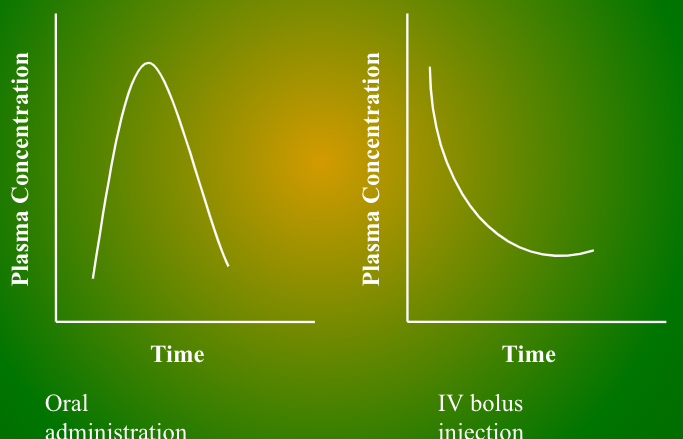
T/F: A single curve typically represents the pharmacokinetics of one dose of a drug (the graph represents oral administration).
True
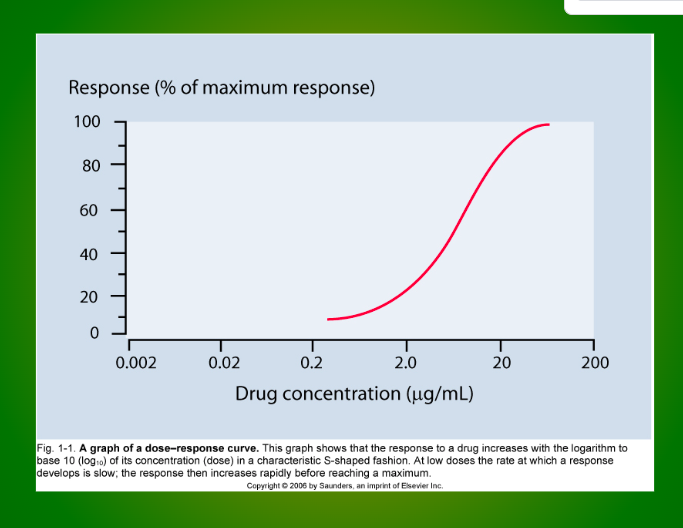
Graded vs. quantal effect curves — difference?
Graded curve = The magnitude (intensity) of a response
Quantal curve = The % of a population that had a response
Is the graded response reversible?
Yes, at a lower dose
Which response is all-or-none?
Quantal response
3 Examples of quantal responses
Toxicity, death, hypotension
Does plateau on conc-effect curve guarantee safe effect?
No, it could be toxic
T/F: Conc-time curve shape is unchanged by dose and same route
True, only Cmax changes
T/F: Higher dose changes half-life
False, half life is constant
Does the Rate of drug elimination increase/decrease/stay the same at higher doses?
The rate stays the same
First order kinetics: The body eliminates a constant fraction of the drug per unit time (ex; 50% per hour)
Therapeutic index (TI)
Toxic dose / Therapeutic dose
T/F: The larger the TI the safer the drug
True
When is response-time curve preferred?
When the plasma concentration does not match the effect
(Ex; It is difficult to measure the plasma levels when taking topicals but the affect can be observed, like reduced inflammation)
4 Reasons why plasma concentration may not reflect the drug effect?
Tolerance; reduced drug response despite same dose
Delays; takes time to equilibrate at the target site
Active metabolites
Chirality; one isomer may be active/inactive
Prodrug
Inactive form of a drug that gets activated inside the body which is usually by metabolism
If an oral dose has a greater effect than IV, what does it suggest?
The oral route activated the prodrug
The drug gets absorbed in the GI tract then metabolized by the liver where liver enzymes converts it from inactive to active
Tolerance vs. Resistance
Tolerance = Host adapts, decreased response to a drug over time
Resistance = Pathogens/cells adapt, drugs become less effective against it (ex; bacteria, cancer cells)
3 Mechanisms of tolerance
Increased Clearance
Decreased Receptors
Decreased Absorption
T/F: Plasma conc. does not reflect response in single-dose therapy
True
Is slow or fast absorption preferred for treating Helicobacter pylori infection?
(H. pylori is a stomach bacteria that can damage stomach and duodenal tissue, causing pain and inflammation.
Slow absorption, it helps keep the drug in the stomach longer
Methotrexate is used to treat cancer and autoimmune diseases. What does its affect depend on?
Duration of exposure not concentration
IV bolus vs infusion
IV bolus = Entire dose is given all at once
Infusion = Steady dose
Why does warfarin take days to work? (Warfarin prevents blood clots)
Clotting factors already in the blood circulation need to degrade
T/F: Fluctuating antibiotic levels may be preferred
True
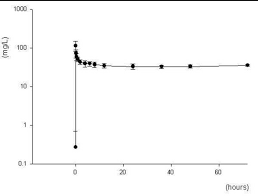
Ceftazidime shows what kinetics and concentration–time curve shape? (Ceftazidime treats bacterial infections)
First order kinetics
Flat curve
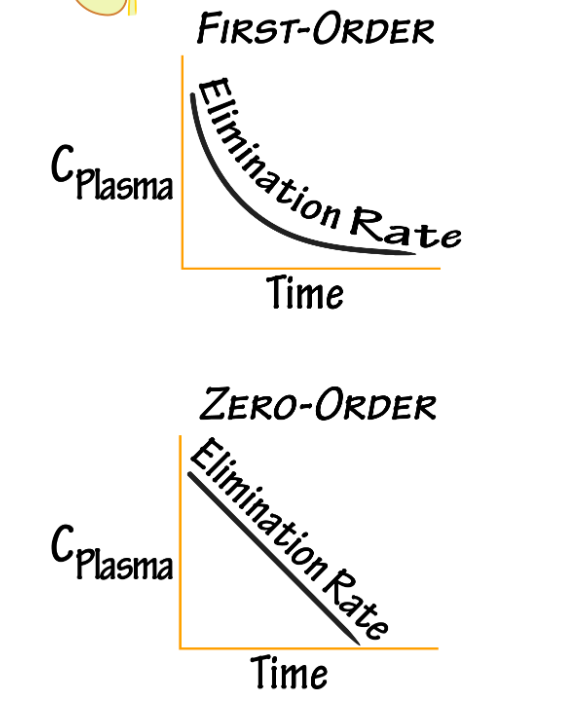
Define First order kinetics. What kind of curve does it have?
The body eliminates a constant fraction of the drug per unit time, so drug elimination increases with concentration (ex; 50% per hour)
Exponential decline curve
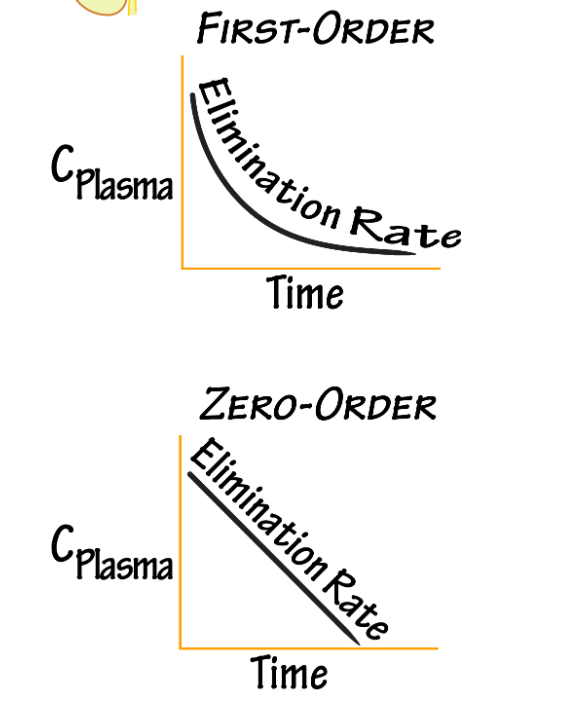
Define Zero order kinetics. What kind of line does it have on a graph?
The body eliminates a constant amount of the drug per unit time (ex; 10 mg per hr)
Linear decline
Which kinetic type is most common in body?
First order kinetics
First-order kinetics equation before and after integration?
Before integration: Rate = -K[C]
After integration: C = C₀e⁻ᵏᵗ
Slope & y-int of log conc-time graph?
Slope = -K/2.303
Y-int = log C₀
How does half-life differ in first order vs. zero order kinetics?
First order kinetics = Constant
Zero order kinetics = Increases with dose
Which kinetics appears linear on semi-log plot?
First order kinetics
What does slope of semi-log plot represent?
Elimination rate
What is the key difference between transcellular and paracellular transport?
Transcellular: Lipophilic drugs passes through cells
Paracellular: Small drugs with low molecular weight (MW) passes between cells, limited by tight junctions
Why does inflammation increase paracellular transport?
It loosens tight junctions, allowing larger/small drugs to pass.
What does paracellular transport depend on?
Molecular weight of the drug (must fit through junctions).
What does transcellular transport depend on most?
Lipophilicity of the drug
Which drugs diffuse best through membranes?
Highly lipophilic drugs.
True or False: Human cells have cell walls.
False — they have cell membranes.
What 3 things can cross the semi-permeable membrane?
Lipophilic drugs
Small hydrophilic molecules
Water
What does the fluid mosaic model describe?
Structure of cell membrane = Fluid lipid bilayer + Embedded proteins for transport
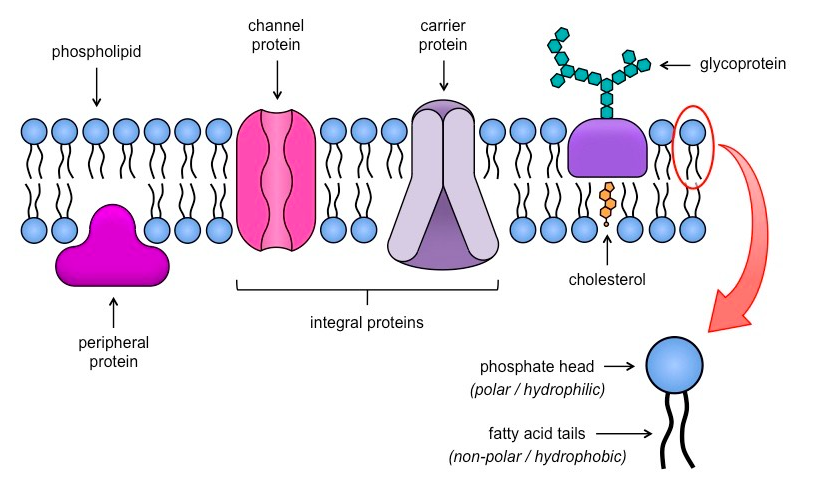
What drives passive diffusion?
Concentration gradient (high → low)
No energy required
True or False: At equilibrium, molecules are equal on both sides (blood vs tissues)
False — only the rate is equal, not the amount.
How can drug absorption be increased at equilibrium?
Increase blood flow to restore concentration gradient
What effects of polycythemia on drug equilibrium and diffusion?
(Polycythemia = Abnormally high RBCs = Thick blood flow)
Increases equilibrium due to less plasma volume
Decreases diffusion due to less blood flow
(which lowers concentration gradient)
What is the simplified form of Fick's Law?
Rate = P × [conc. at absorption site].
Why is Cp (plasma concentration) negligible in Fick's Law?
Absorption site concentration > Plasma concentration
Do lipids follow Fick’s law?
(Fick’s law: Rate of drug diffusion across a membrane)
No — they diffuse freely.
What order of absorption does Fick’s Law describe?
First-order absorption, Higher concentration = Faster absorption rate
Why is absorption usually faster than elimination?
There is a larger concentration gradient between the absorption site (e.g. gut) and the blood
Most drugs are weak. What is the implication?
Weak electrolytes — they are both hydrophilic and lipophilic.
What is the solubility difference between ionized and unionized drugs?
Ionized = Water-soluble
Unionized = Lipid-soluble
Where do weak acids accumulate — stomach or plasma? Why?
They get ionized in plasma so they accumulate
They remain unionized in the stomach so they get absorbed
Where do weak bases accumulate and why?
In the stomach — they become ionized there = ion trapping
Key Concept: "Like absorbs like"
Acidic environment (e.g. stomach): favors _____ → better absorption
Basic environment (e.g. intestines): favors _____ → better absorption
Unionized weak acids
Unionized weak bases
Which pharmacokinetic phase is influenced by protein binding?
Distribution.
What is the effect of plasma protein binding on drug half-life and tissue affinity?
Increases half life
Decreases tissue binding
How does tissue binding affect half-life?
It decreases half-life — the drug is pulled into tissue.