Lecture #8 | Tree Building
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
33 Terms
Discrete characters
No overlapping variation
teeth, no teeth
Divided into binary or multistate
Binary (0 or 1)
Multistate ( >2 states, 0 or 1 or 2)
Continuous characters
Measurements
Multistate characters
May be ordered (linear) or unordered
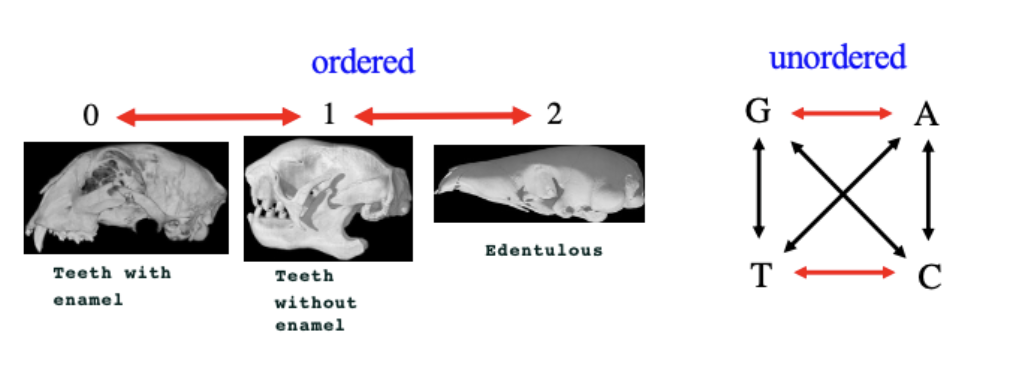
How are numerical scores assigned?
Usually (not always), the character state considered to be most ancestral is given the lowest numerical value, with more derived states given an increasingly higher value (1,2…) depending on the type of character
usually assigned with an initial hypothesis of character change developed by the investigator
Subject to reevaluation
Character polarity
Assignment of character order
evolutionary history of a trait or feature of an organism
Outgroup method
A method to determine polarity
If a character has 2 or more states, the state found in the next most related group (outgroup) is ancestral
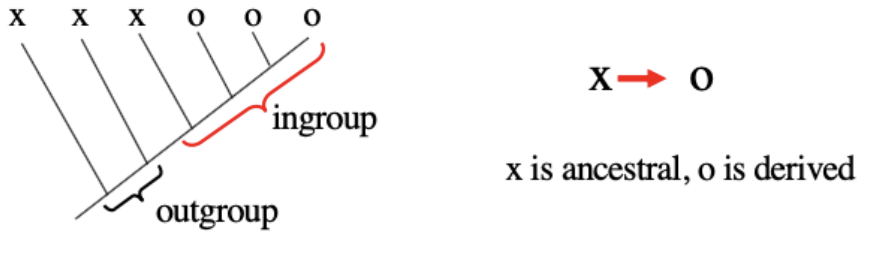
By far the most common tree rooting method
Best to have two outgroups because it defends against autapomorphy (unique change in character state that is not informative for relationships)
What is important when considering methods of analysis for a tree?
Informative-organized
Predictive-Provides information that is fundamental to the relationships
Stable- stable to new info from new taxa and data
Operational- based on a set of procedures that can be accepted and refuted
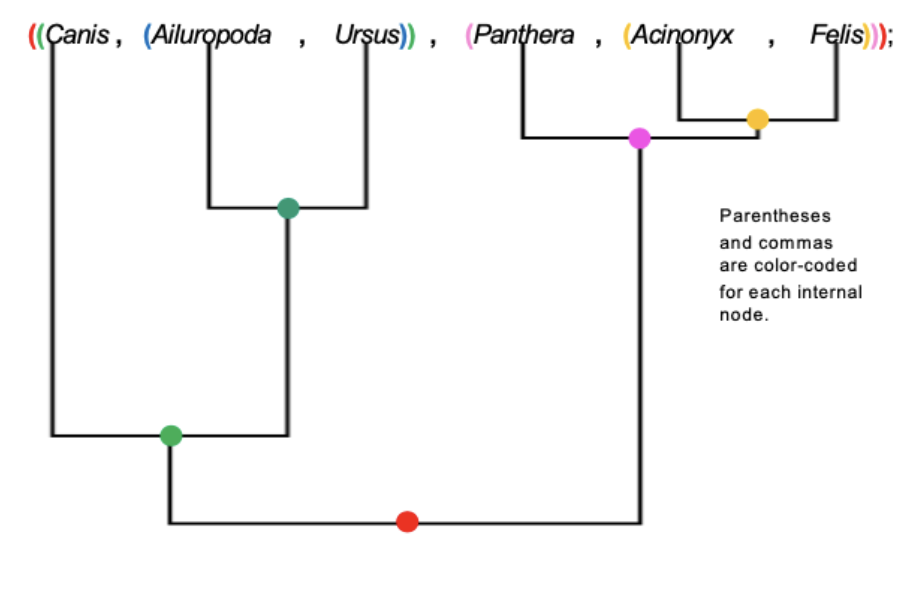
Newick Format
Method to represent graphical trees, with or without branch lengths, using parentheses, commas, and a semicolon
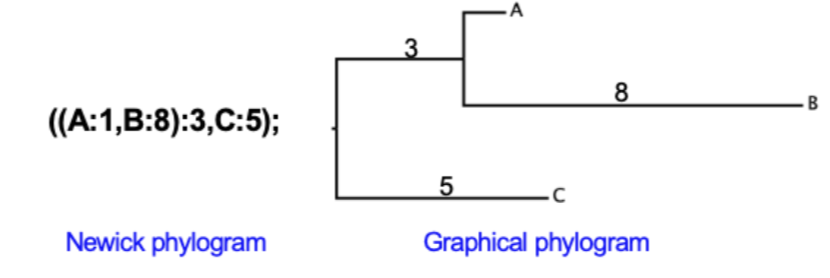
How to construct a newick tree
Identify internal nodes
Add a set of parentheses for each internal node and a comma between the left and right descendants of each nodes. Add a semicolon to the end of the Newick tree
Each branch length is preceded by a colon (;)
Branch lengths (with their preceding :) are placed after the taxon names and after right parentheses (except the last one)
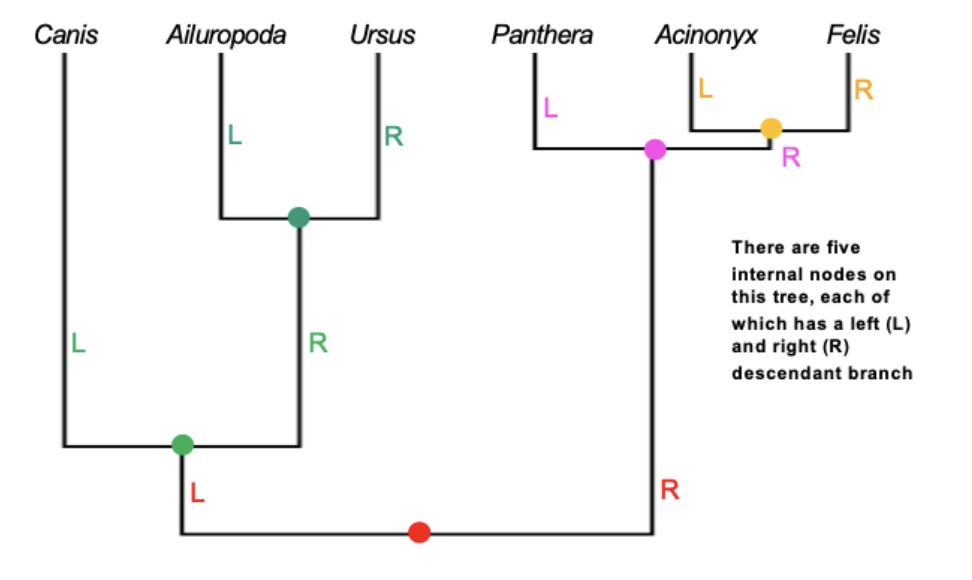
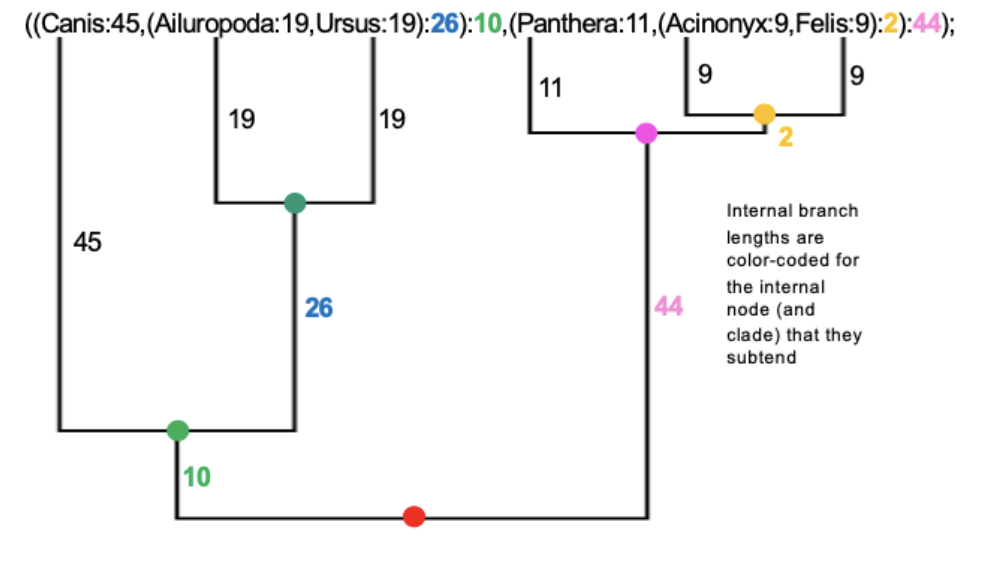
Binomial names require apostrophes or underscores to link together

Unrooted trees
Lack temporal polarization
point of common ancestry is missing
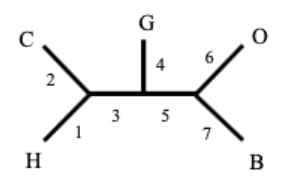
Rooted trees
Temporally polarized
point of common interest is given
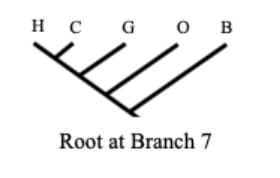
Determining unrooted number of taxa
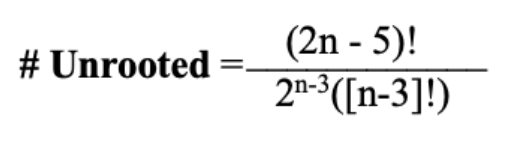
where n = number of taxa
Determining number of rooted trees
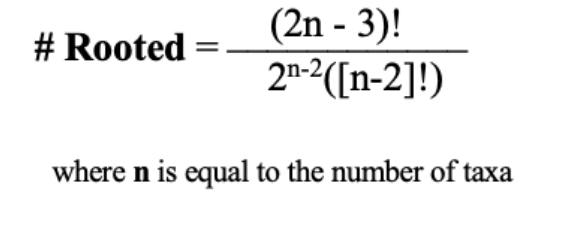
Gene duplications root
Paralogous gene duplications that predate the common ancestor of a taxonomic group are used to root the tree
root is placed between paralogous gene copies
Midpoint rooting
Tree is rooted on the midpoint between the two most distant leaves
choose the midpoint between the two most distant external nodes
assumes the rate of evolution is the same on the longest branches of the tree
Desirable properties of tree building methods
Consistency: will the method converge on the correct solution given enough data
Efficiency: How fast is the method
Power: How much data is needed for a reasonable result
Robustness: Will minor violations of the assumptions result in poor estimates of phylogeny
Types of data: Discrete versus Continuous
Discrete data is more common-few methods can handle continuous data
Types of data: Character versus Distance Data
Important that character comparisons between taxa can be used to develop distance matches, but reverse cannot happen
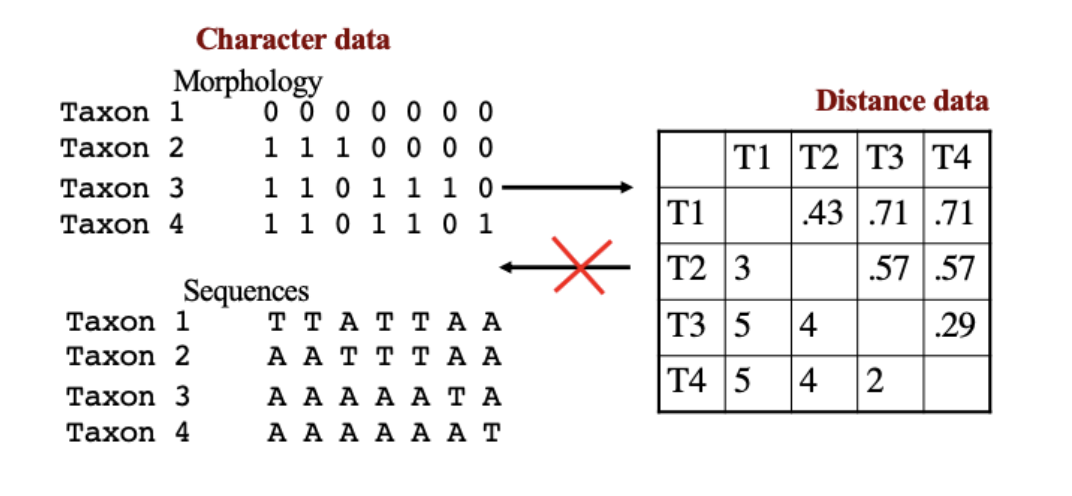
Types of data: Correct versus uncorrected data
Morphological characters may be standardized so that they all have equal value in an analysis
Cluster analysis
The recognition of groups of individuals on the basis of multiple characters. Groups may be mutually exclusive, hierarchic, or partially overlapping
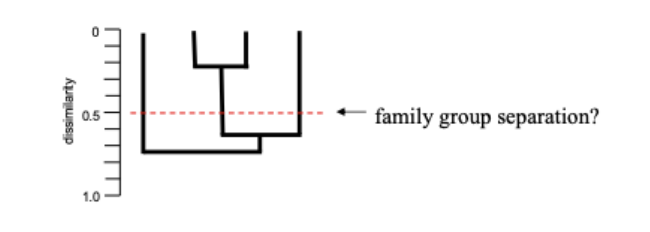
Phonetics
classification based on numerous precisely delimited characters of equal weight and their comparison by an explicit method of grouping
Key points of cluster analysis
Objectivity
Polythetic Taxa: Groups based on character combinations
Many characters- use as many characters as possible
Equal weighting-every character has equal weight
Overall similarity: groups recognized on basis of overall similarity nothing else
Defining character polarity is nor important
How to create a cluster analysis
Select taxa that represents both the entire geographical range and the entire morphological range of variation
Select characters: As many characters as possible should be chosen. Each character gives equal weight to the determination of overall similarity
Calculation of similarity/dissimilarity matrix
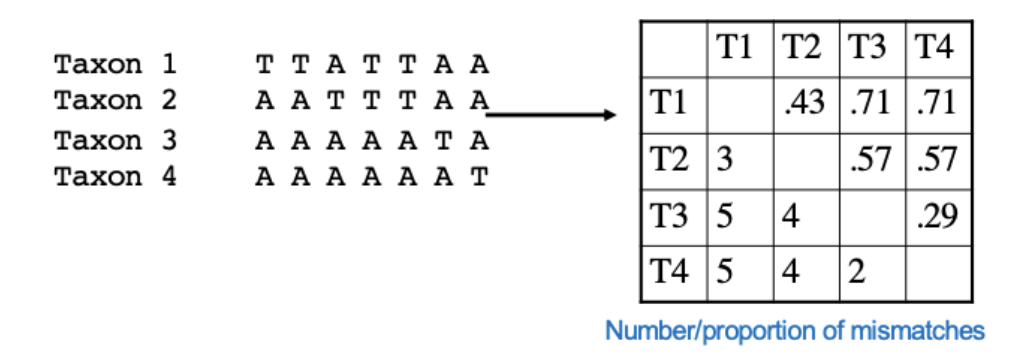
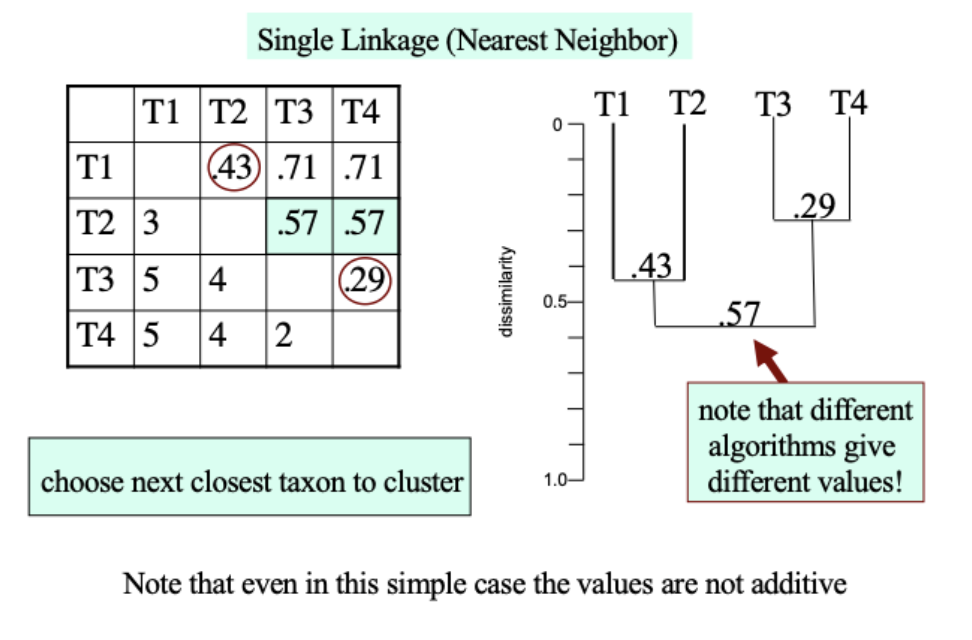
Grouping OTUs by single linking (nearest neighbor)
Advantaged of cluster methods for determining relationships
Operational: clearly defined procedures
Communicable: anyone can code for characters and produce a classification without prior knowledge
No weighting or preference for certain characters
Problems of cluster methods
Relationships depicted are strongly affected by
choice of characters
number of taxa
type of similarity coefficient
Clustering technique applied to similarity matrix
higher categories are subjective
Groupings are more technique dependent providing an artificial grouping of taxa rather than moving towards a system of uncovering stable relationships
How often are phenetic (cluster) methods used?
Rarely do studies that utilize morphological characters ever use phenetic methods.
However, phenetics are still used for relatively simple organisms like prokaryotes
What assumptions allow for use of distance methods for molecular data?
Molecular clock is assumed whereby mutations at any particular site in the genome are random and occur with equal frequency over time
most changes are observed
Character system is enormous with a potential to use the entire genome for analysis
Changes in the genome are expected to be independent of environmental or selective pressure and less subject to convergence
But we know
There is a preference for transitions
not all positions of codons change at equal rates
different regions of the genome,e are more conservative
Morphometrics
The quantitative description, analysis, and interpretation of shape and shape variation in biology
Single linkage clustering method
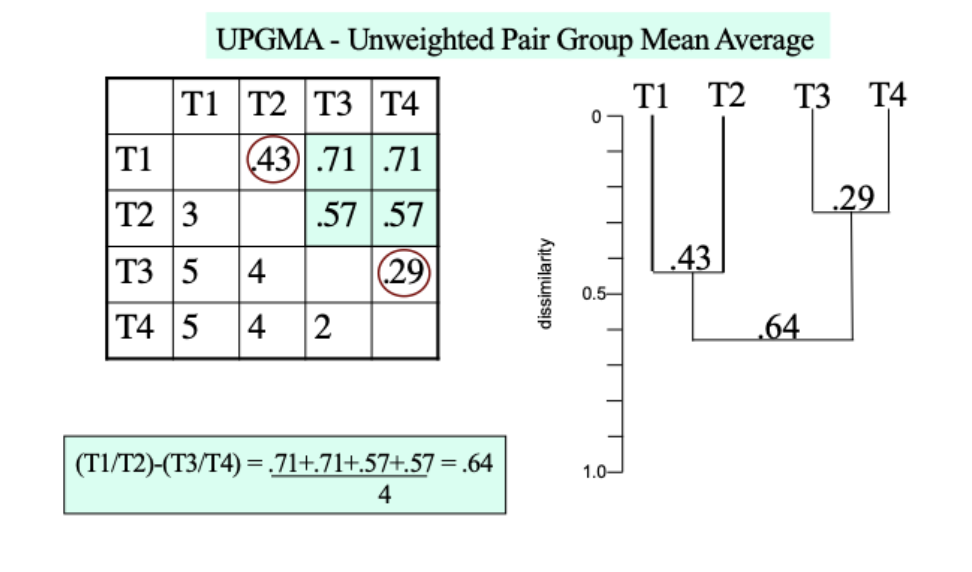
UPGMA- Unweighted pair group mean average
Neighbor-Joining Methods
Widely used for constructing phylogenetic trees with molecular data
Can be applied to the data for the corrected matrix conversion
Assumes additivity, not ultrametricity, so all branch length divergencies are not necessarily equal
Branch lengths in the matrix and the tree path length match perfectly and there is a single and unique additive tree that fits the distance matrix
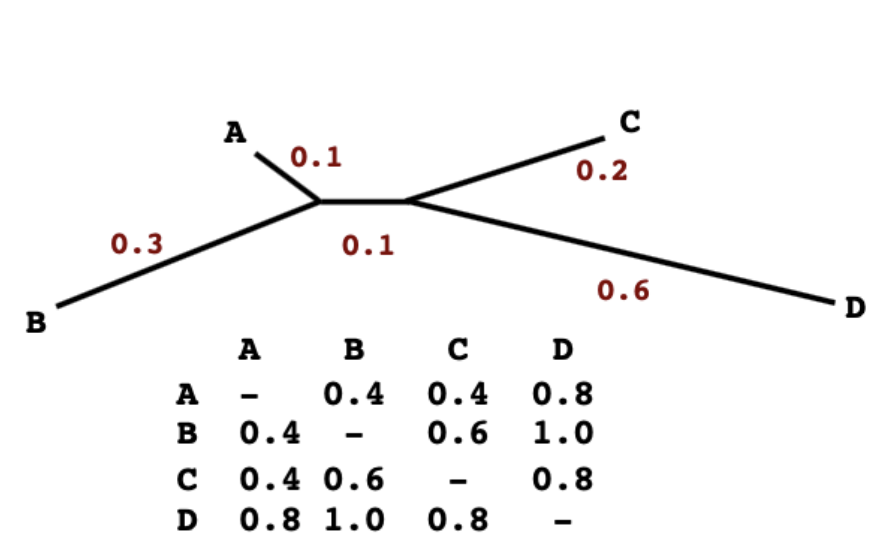
Advantaged of NJ
Branch lengths are additive and reflect the true distances between taxa
Fast computational time
Can invoke outgrip rooting of the tree
Can empty various models of character state evolution to adjust branch lengths relationships
Disadvantages of NJ
not possible to infer or directly map character back onto topology
produces a single tree with no evaluation of competing hypotheses
can produce a quick and dirty tree that may be very different from OC method trees