The LADMER System: Metabolism
1/105
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
106 Terms
Drug Metabolism
The irreversible conversion of a drug to a different substance in vivo by enzymatic catalysis or biochemical transformation of drug to metabolic products
Liver
Principal/Main site of metabolism
●Lung - for inhalation/volatile gases
●Skin
●Gastrointestinal mucosal cells
●Microbiological flora in the distal portion (part na nagb-biotransform) of the ileum
●Large intestine
●Kidneys
Other/Minor sites of metabolism
True
T or F
The decline from peak plasma concentrations after drug administration results from drug elimination or removal by the body.
First-Order Elimination
The metabolism rate constant (km) is the sum of the rate constants for the formation of each metabolite.
Rate Constant (k)
rate constant of elimination
kR or ke
first-order rate constant for excretion
kNR or km
first-order rate constant for metabolism
○ METABOLISM (Biotransformation)
○ EXCRETION (Renal)
How are drugs removed from the body?
Liver
-Major organ responsible for drug metabolism
-Both a synthesizing and an excreting organ
Liver lobule
-Basic anatomical unit
-contains parenchymal
cells in a network of interconnected lymph and blood
vessels
hepatic artery
carries oxygen into the liver (~25% of blood supply)
Hepatic Portal Vein
Carries nutrients to the liver (~75% of blood supply)
Hepatic Vein
Drains deoxygenated blood from the liver into the IVC (inferior vena cava)
Common Bile Duct
Drains bile and biliary excretion products from both lobes into the gallbladder
Sinusoids
-Large vascular capillaries where the terminal branches of the hepatic artery and portal vein fuse within the liver
-Facilitates drug and nutrient removal before the blood enters the general circulation
Sinusoids
Lined with endothelial cells of Kupffer cells (phagocytic tissue macrophages that are part of the RES (Reticuloendothelial System); engulf worn-out RBCs and foreign material)
flow and site
Drug metabolism in the liver has been shown to be ____________ dependent
-Hepatic diseases
-genetic differences in enzyme levels
-environmental factors
What can affect half-lives of drugs eliminated by drug metabolism
Drug Biotransformation
-Drugs are chemically converted in the body to metabolites
-Usually an enzymatic process
1. Deactivation
2. Activation (Prodrugs)
3. Active drugs with active metabolites
4. Active drugs with toxic metabolites
POSSIBLE EFFECTS AFTER METABOLISM:
Prodrugs
Pharmacologically inactive compounds designed to maximize the amount of the active species that reaches its site
Enalapril
antihypertensive (ACEi)
Dopamine
for Parkinson's Disease
Diazepam
Long-acting sedative used for muscle spasms.
Demethylation
Metabolic process converting Diazepam to Desmethyldiazepam.
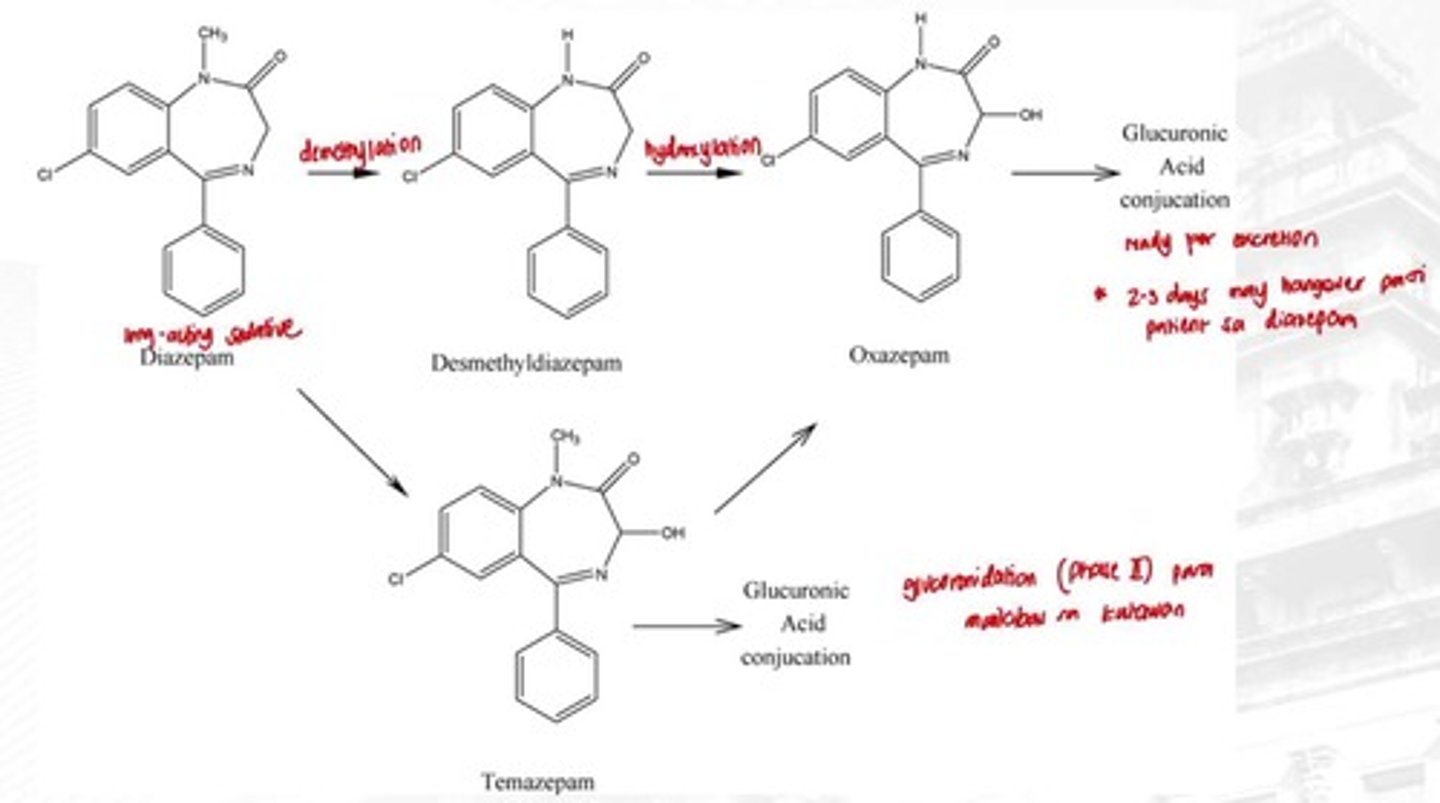
Hydroxylation
Conversion of Desmethyldiazepam to Oxazepam.
Active Metabolites
Metabolites that retain pharmacological activity.
Toxic Metabolites
Harmful metabolites produced during drug metabolism.
First-Pass Effect
Reduction of drug concentration before systemic circulation.
Drug metabolism
Conversion to a more excretable form (more water-soluble form)
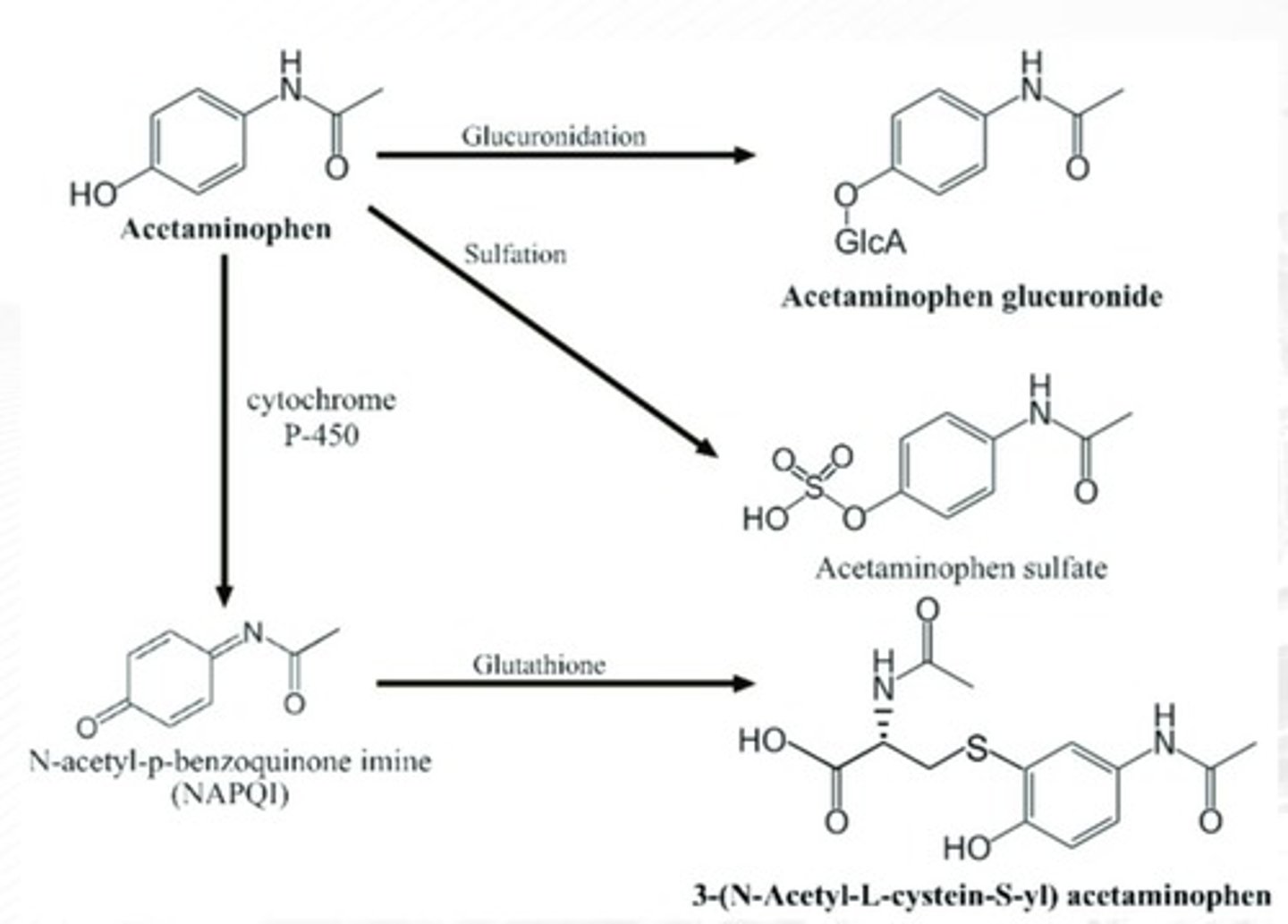
NAPQI
Toxic metabolite causing liver damage if there's not enough GSH in the liver
Mixed-function oxidases (MFOs)
Hepatic enzymes that are responsible for oxidation and reduction of drugs (xenobiotics), and certain natural metabolites (ie. steroids)
MFOs are contained in the parenchymal cells of the liver in association with the endoplasmic reticulum (a network of lipoprotein membranes within the cytoplasm and continuous with the cellular and nuclear membranes)
MFOs are contained in the ______ of the liver in association with the _______ (a network of lipoprotein membranes within the cytoplasm and continuous with the cellular and nuclear membranes)
converts lipophilic substance -> hydrophilic
Main mechanism (MFOs)
Phase I
-mostly REDOX & hydrolysis reactions
● Asynthetic reactions
● Oxidation
● Reduction
● Hydrolysis
PATHWAYS OF DRUG BIOTRANSFORMATION:
Phase I
● Synthetic reactions
● Conjugations
PATHWAYS OF DRUG BIOTRANSFORMATION:
Phase II
Phase II
-Conjugation reactions
-To make drug more ionized (and readily excretable)
Phase I
-Functionalization
-To make drug more polar, to increase excretion in urine
CYP450
-known metabolic enzyme identified by 450nm wavelength
-has a lot of cytochrome isozymes
Phenylbutazone
mostly used for rheumatoid arthritis;
not commonly used since it has a lot of ADRs
CYP3A4
most abundant in the liver; causes first-pass effect
CYP2E1
for alcohol metabolism
False
IV drug will not undergo Phase I and Phase II; straight to elimination
T or F
IV drug will undergo Phase I and Phase II; straight to elimination
PHASE I - OXIDATION
-Addition of Oxygen or negatively charged radical
-Removal of Hydrogen or positively charged radical
PHASE I - OXIDATION
What Phase?
-AROMATIC HYDROXYLATION
-OXIDATIVE DEALKYLATION
Phase 1 - Reduction
Phase 1 Reaction:
-Addition of Hydrogen or positively charged radical
-Removal of Oxygen or negatively charged radical
Methylation
Phase II reaction adding methyl groups to drugs.
Acetylation
Phase II reaction removing carbonyl groups from drugs.
Phase 1 - Hydrolysis
Phase I reaction:
-drug is split combining with water molecule
-carried out by hydrolytic enzymes
forms a variety of polar functionality susceptible for
Phase II metabolism
PHASE II - GLUCURONIDATION
Phase 2 Reaction:
-Most dominant conjugative pathway
-Conjugating agent: Glucuronic acid
Uridine diphosphoglucuronic acid (UDPGA)
High-energy intermediate in Phase II Glucuronidation
Glycine;
Coenzyme A thioesters
PHASE II - GLYCINE CONJUGATION
-Conjugating agent ____
-High-energy intermediate: ____
PHASE II - GLUTAMINE CONJUGATION
Phase II reaction:
-Conjugation with coenzyme A (CoA) followed by conjugation with amino acids such as taurine and glutamine.
Sulfotransferases (SULTs)
Conjugating agent in PHASE II - SULFATION
PHASE II - METHYLATION
In humans, drugs and xenobiotics can undergo O-, N-, and S- methylation catalyzed by methyltransferases.
PHASE II - ACETYLATION
-The acetylated product is usually less polar than the parent drug.
-The less polar metabolite can be reabsorbed in the renal tubule and has a longer elimination half-life.
N-acetyltransferase
-enzyme responsible for catalyzing the acetylation of hydralazine (for hypertension), isoniazid (anti-TB), procainamide, and other drugs demonstrates a genetic polymorphism
-"Slow inactivators" and "rapid inactivators"
Glutathione (GSH)
-a tripeptide glutamic acid-cysteine-glycine.
-important in the detoxification of reactive oxygen intermediates into nonreactive metabolites.
N-acetylcysteine
-Antidote for APAP poisoning
-a drug molecule that contains available sulfhydryl (R-SH) groups.
Mixed-Function Oxidases (MFOs)
-Monooxygenase enzymes
-Responsible for redox of drugs and natural
metabolites
CYP isoenzymes
-Catalyzes the biotransformation of various
endogenous compounds (Steroids)
-Located in other tissues - kidney, GI tract, skin,
lung
Reactive Oxygen Species (ROS)
Chemicals causing advanced aging.
CYP2B6
Human isozyme that metabolizes antidepressant drugs
CYP2C9
Responsible for warfarin metabolism in humans.
CYP2C19
Metabolizes proton pump inhibitors (PPIs) like '-prazoles'.
CYP1A2
Metabolizes methylxanthines such as caffeine.
CYP3A4
Most abundant CYP enzyme in the liver, causes first-pass effect
CYP2E1
Isozyme for alcohol metabolism
Alcohol- and Aldehyde- dehydrogenase
-Found in the soluble fraction of liver
-Involved in metabolism of ethanol
Xanthine oxidase
Converts hypoxanthine xanthine to to uric acid in metabolism.
Xanthine oxidase inhibitors
Lower serum uric acid; used for gout treatment (anti-gout)
Enzyme inhibitors
drug/chemical that reduces enzyme activity
Reversible inhibition
increasing the dose of the second drug taken can displace the first drug that was administered.
Competitive inhibition
Inhibitor competes for the same enzyme binding site; mechanism is direct and is rapidly reversible
Noncompetitive inhibition
Inhibitor and substrate do not compete for the same active site, because of an allosteric site.
Irreversible inhibition
permanent inhibition of the enzyme
First-pass effect
A phenomenon where a portion of the orally administered drug undergoes elimination before it has a chance to be absorbed into systemic circulation.
First-pass effect
Presystemic Elimination
Enzyme inducers
drug/chemical that increases enzyme
activity by transcriptional activation leading to increased synthesis of more CYP enzyme proteins.
Intrinsic clearance
-The ability of the liver to remove drug independently of blood flow
-Primarily due to the inherent ability of biotransformation enzymes to metabolize drug as it enter the liver
-Presence of other drugs
-Presence of environmental condition
Intrinsic clearance depends on:
Fractional difference
the extent to which an orally administered dose undergoes first-pass effect
1. The entire drug dose administered orally is released from dosage form and is made available for absorption,
2. The drug is not acid-labile
3. The drug is not destroyed in the GIT by the GI enzymes
4. The AUCoral < AUCIV
What conditions must be met, in order for a drug to undergo first-pass effect?
Increased drug effect
Higher plasma concentration leading to stronger effects.
False
Drugs with Fractional difference > 0.9 are best administered by routes other than those involving the GIT
T or F
Drugs with Fractional difference < 0.9 are best administered by routes other than those involving the GIT
Hepatic clearance
-Volume of drug-containing plasma that is cleared of the drug by liver per unit
-Includes excretion of drug into the bile
-Drugs with relatively high MW (>500)
-Polar drugs
-Glucuronide conjugates of various drugs
Biliary excretion involves:
Hepatic Extraction Ratio
-The fraction of the drug removed from plasma by the live
-Amount of drug removed from plasma by the liver
Hepatic Blood Flow (QH)
Rate of blood flow through the liver.
INCREASED QH, INCREASED rate of removal of drug by the liver
INCREASED QH, _____ rate of removal of drug by the liver
DECREASED QH, DECREASED rate of removal of drug by the liver
_____ QH, DECREASED rate of removal of drug by the liver
~1.5 L/min.
hepatic blood flow under normal conditions: ___
○ Presence of drugs (Example: Propranolol)
○ Presence of disease state
○ Exercise
Hepatic blood flow may be affected by:
Enterohepatic Circulation
-Reabsorption of the drug after hepatic pass
-Recycling of drug
CA
the arterial concentration of drug in plasma entering the liver
CV
the venous concentration of drug in plasma exiting the liver
may range from 0 to 1
Hepatic Extraction Ratio Range
HER < 0.2 indicates low drug removal.
Low Hepatic Extraction Ratio