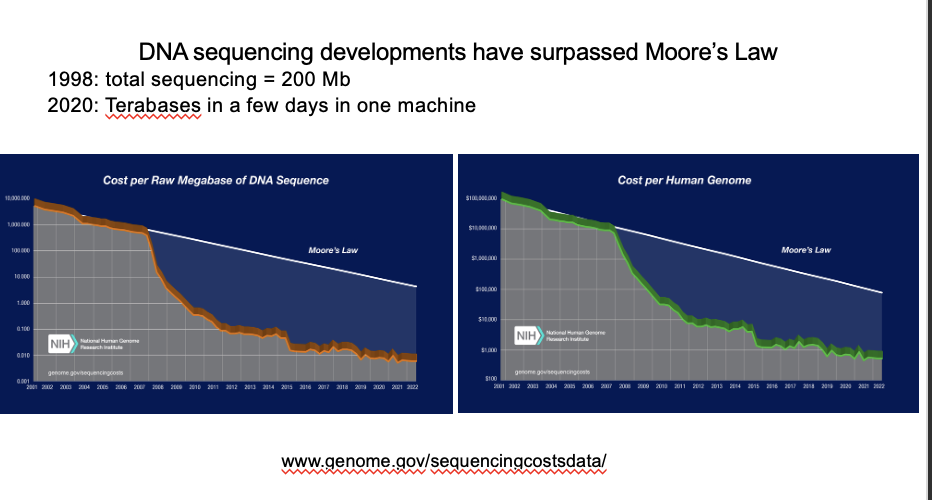
L2: Applying massively parallel sequencing detecting and interpreting genome variation
1/50
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
51 Terms
Applications of massively parallel sequencing
Making de novo genome sequencing feasible
Sequencing cancer cell genomes
Sequencing clinical isolates to identify causative pathogens
Metagenomics
Archaeological genomics
Phylogenomics
RNA-seq
Making de novo genome sequencing feasible
combining PacBio (long reads) with Illumina (short reads, higher throughput)
with
Paired end reads
Sequencing cancer cell genomes
to identify changes likely to be
causitive, monitor cancer progression and tumour ecosystem
classify cancer for therapy
Sequencing clinical isolates to identify causative pathogens
tracing their spread by their nucleotide divergence
e.g: → how new SARS-CoV2 variants have been identified and tracked
if no infrastrcuture to courier large numbers of samples from the field to sequencing centres
→→portable nanopore sequencing allows speed and portability
Metagenomics
Sequencing of ecosystems
e.g soil, sea, gut contents
Single-molecule-based sequencing potentially yields a ‘metagenome’
i.e parasites, viruses, bacteria that colonise the host tissue from which DNA is extracted
note: single-molecule sequencing is highly sensitive to contamination by DNA of researchers, samplers etc
Archaeological genomics
on minute amounts of DNA
e.g Neanderthals
Phylogenomics
e.g
10,000 vertebrate genomes, including distant branches and endangered species→ 260 to date
70,000 eukaryotes in GB 12 marine worms, 11 lepidotera, 10 mountain bryophytes, 9 Oxfordshire earthworms, 8 diverse diptera, 7 types of apple, 6 algae cultures, 5 festive fucoids, 4 fungi, 3 coastal lichens, 2 chordates
GOAL→ 1.5 M known eukaryotes over 10 years

RNA-seq
profiling gene expression by sequencing reverse-transcribed RNA populations
Population genome sequence projects: cost of sequenceing large numberes of individuals
although gradually decreasing
still expensive
Population genome sequence projects:
1000 genomes consortium
UK10K consortium (2015)
NHS England Genomics England

1000 genomes consortium
after this came a number of 10k genome consortia
now NHS england has a 100k genome consortium
there are others worldwide
UK10K consortium (2015)
identifies rare variants in health and disease
NHS England Genomics England
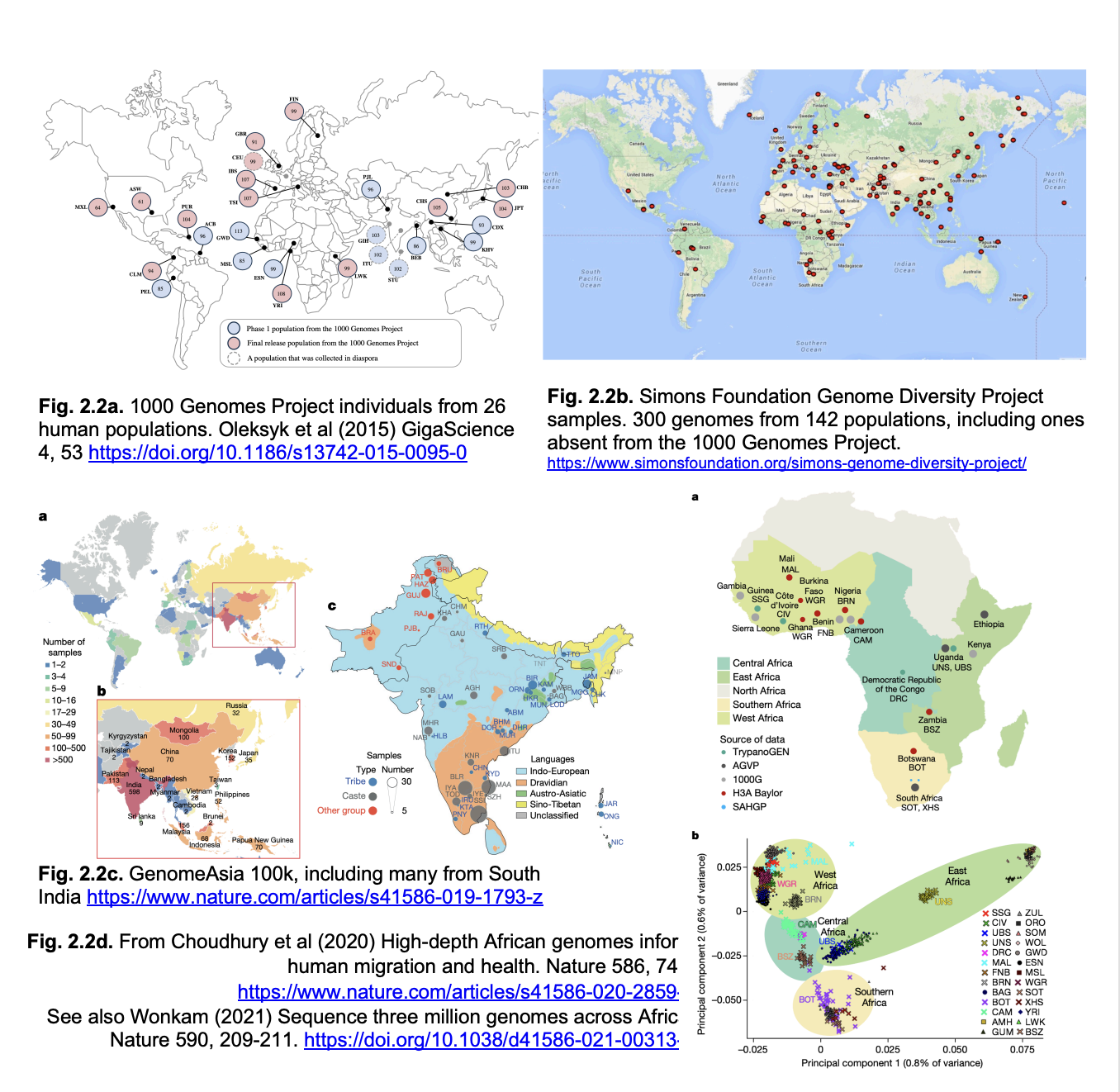
100,000 genomes
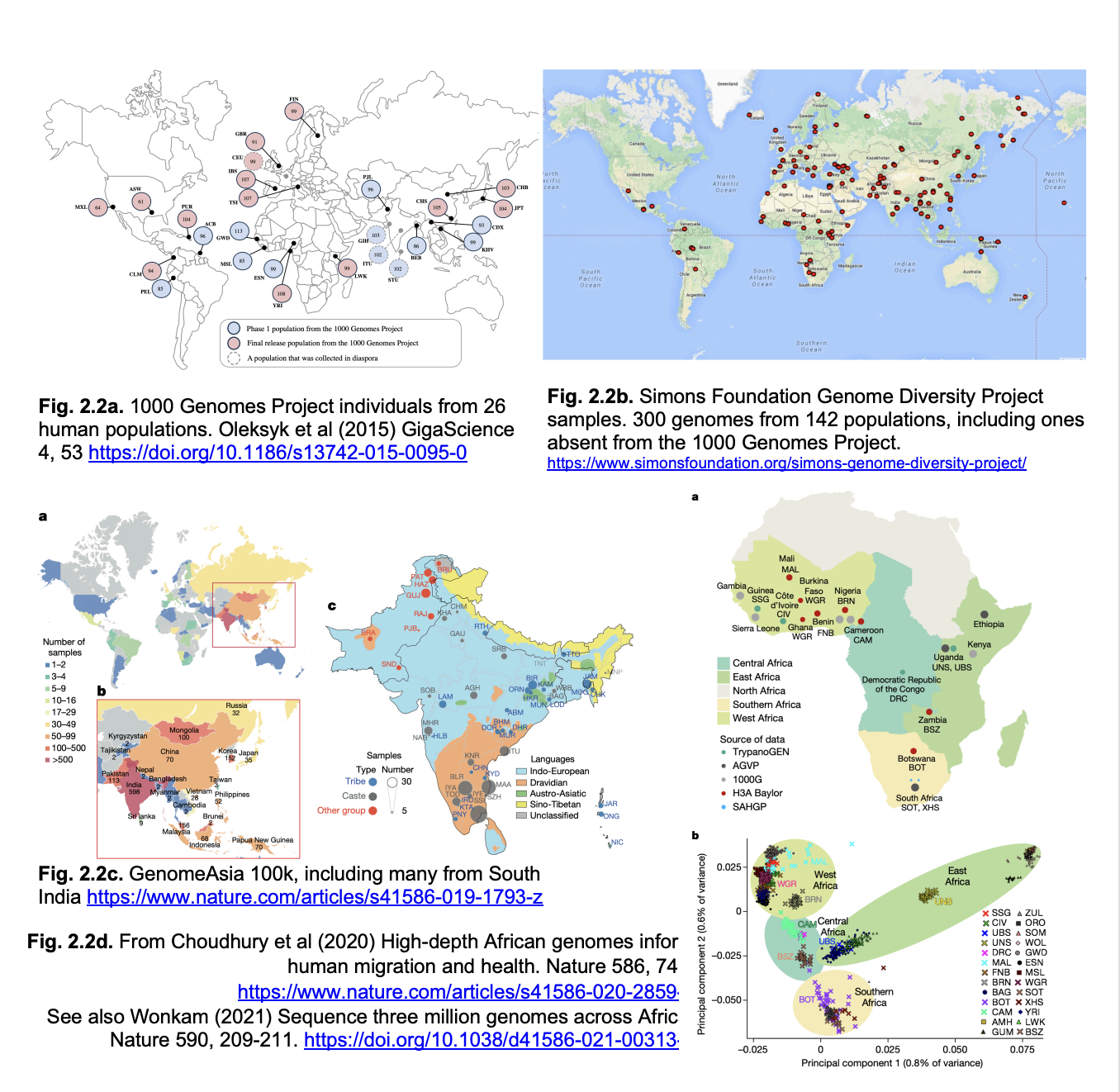
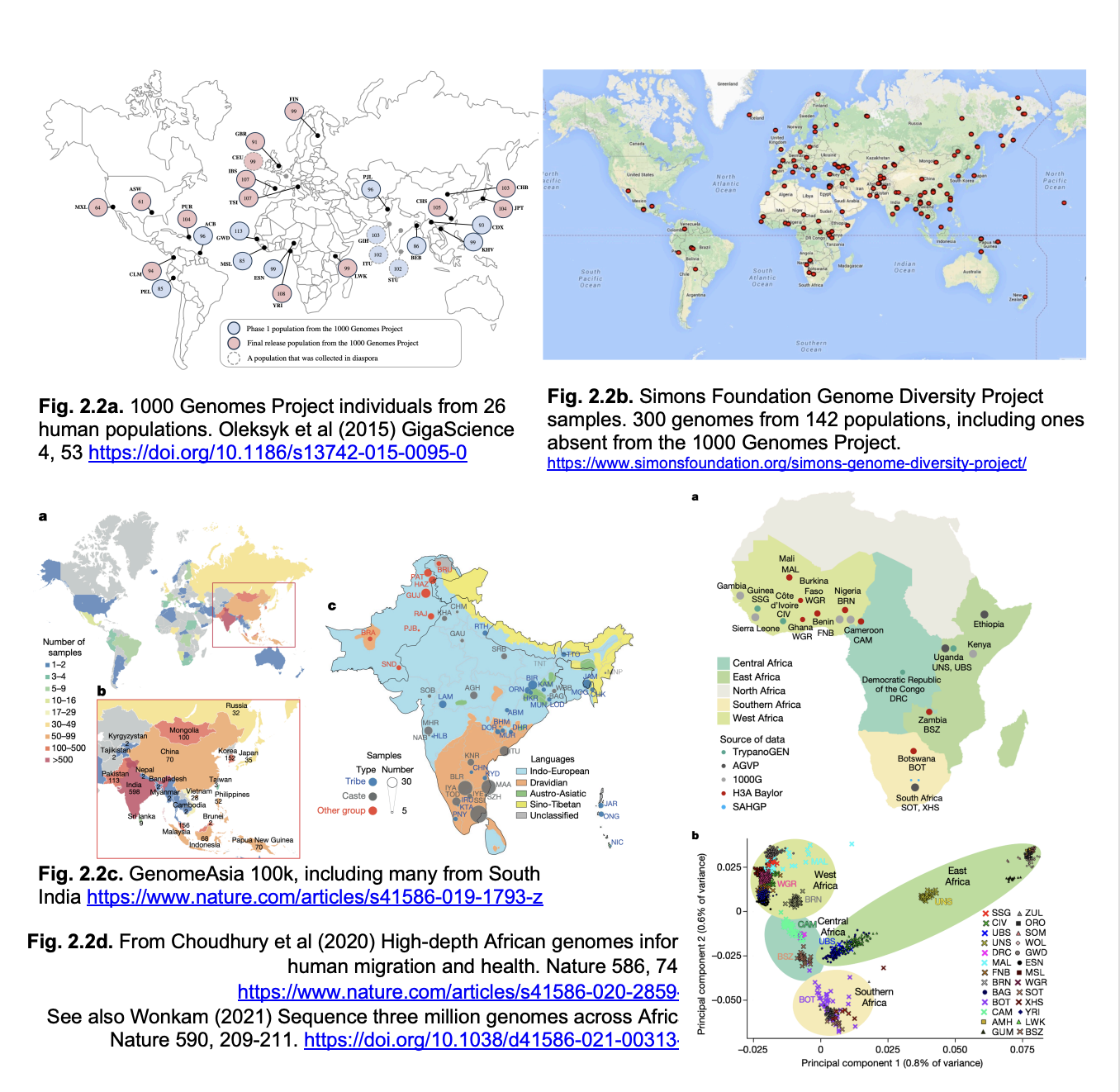
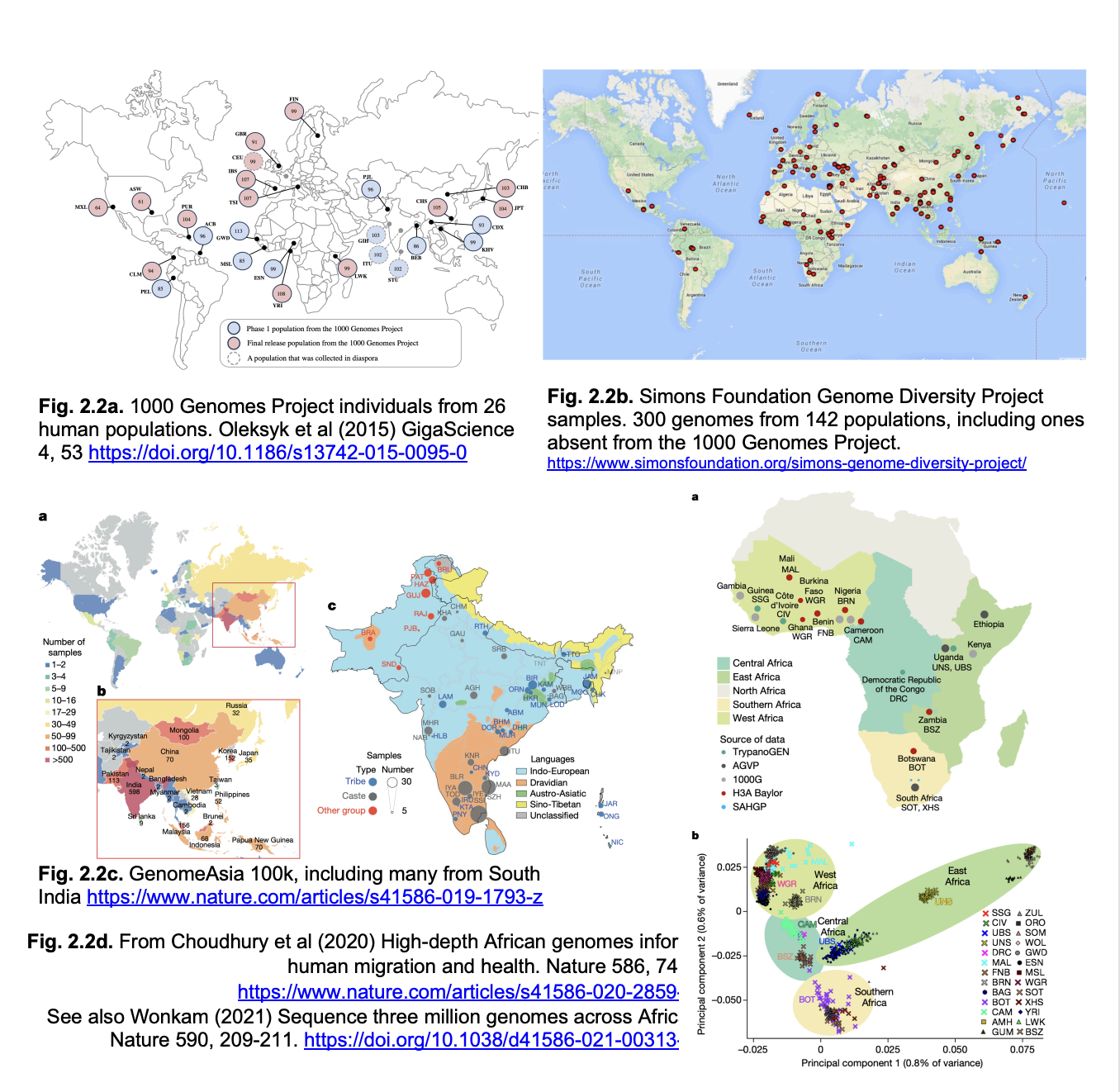
Genomes represented
over-representation of white/European genomes
misses the full range of human genetic variation
especially in Africa where modern humans originated
and where genetic diversity is highest
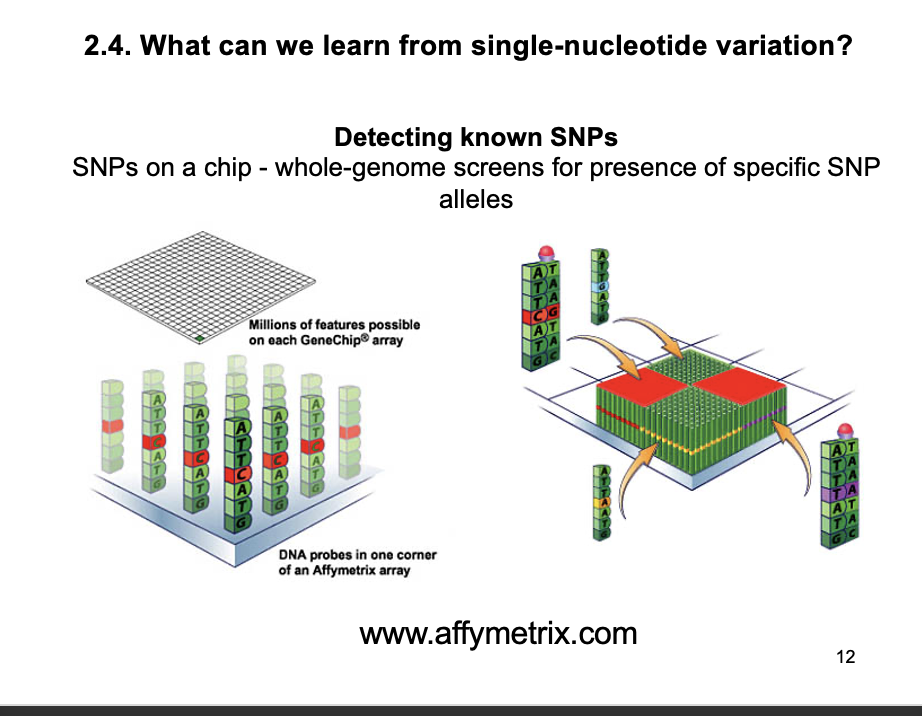
Single nucleotide polymorphisms: SNPs→ how to find
single next-generation sequencing uses single molecules
heterozygous SNPs appear as different bases in different reactions

What are polymorphisms
alleles present at a frequency that is too high to be recent mutation
before elimation by negative selection
→ rule of thumb is a frequency of >1%
Alleles that are rarer than this may be…
recessive lethals
→ will eventually be eliminated
(rare SNPs)
Alleles at higher frequency…
more likely to be selectively neurtral
their frequency can incrase or decrease by ‘genetic drift’
(rare advatageous SNPs)
SNPs causing amino acid substiution
about 1/300
→ most are silent and probably neutral

By seqeuncing many individuals
can make extensive SNP catalogs
but
may still miss SNPs from ethnic groups not sequenced
e.g African populations stil undersmapled!
e.g: 150000 human genomes from diverse UK populations reveal:
600M SNPs
(1 nucleotide in 5 in the genome)

SNP catalogs from diverse populations reveal…
highest variation (heterozygous sites per kb) in AFRICA
Decreasing with distance from Africa
THEREFORE: supporting the region as the one where modern humans originated
(or at least most of their genomes)
Origin of SNPs
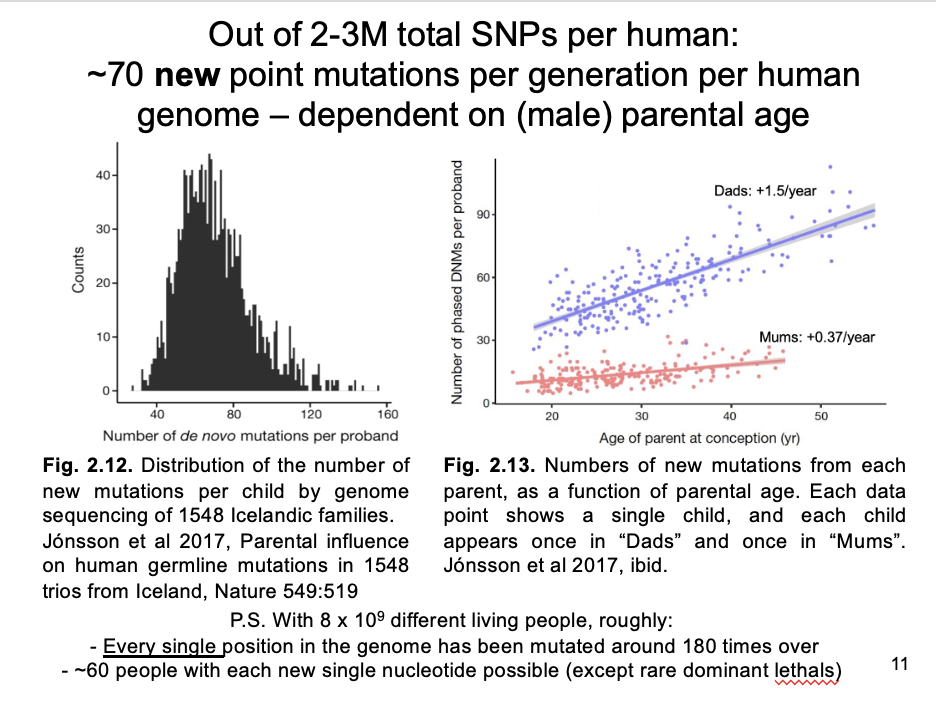
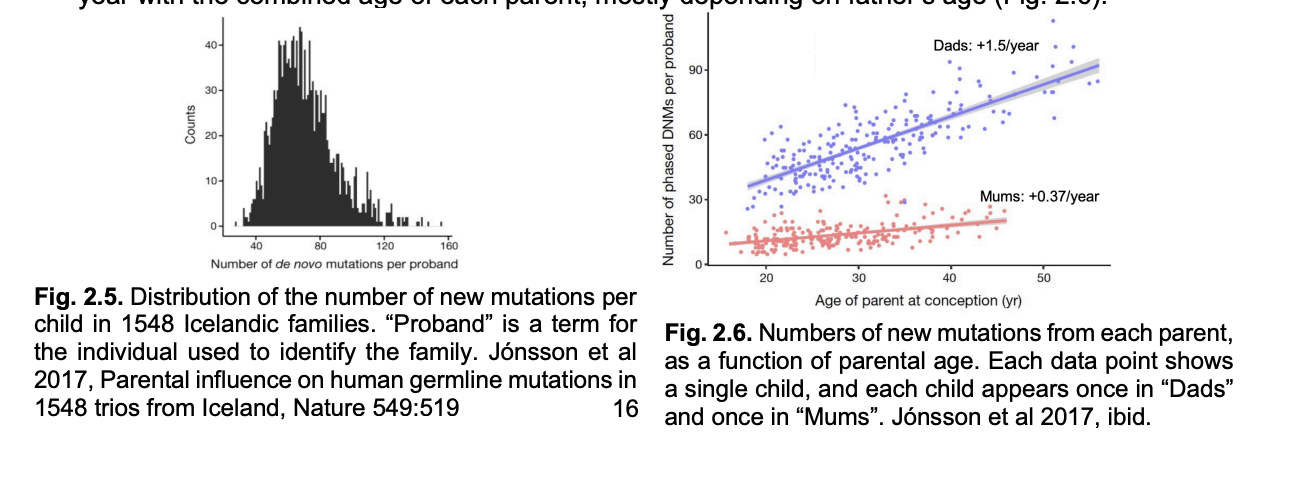
population sequencing of human pedigrees suggests a mutation rate of 1.3 × 10^-8 nucleotide substitutions per bp per gen
Multiple mutation rate per bp with size of the human diploid genome
→ 70 new mutations per generation per diploid genome:
this is a tiny % of the 2M-3M SNPs in most humans (number estimated from heterozygosity)
BUT: a continual source of new variation
Note:
With 8 billion humans on the planet, their pan-genome carries ~560 billion new mutations per generation
i.e. each basepair in the genome is mutated on average ~180 times globally per generation
Mutation rate male vs female germline
x4 higher in male than female germline
increases by about 2 mutations per year with the combined age of each parent
→ mostly depending on father’s age
What can we learn from single-nucleotide variation?: Large-scale SNP detection
i.e post-discovery by hybridisation
calssical genetic mapping identifies rare high-risk disease alleles
if pedigrees are available
BUT: poor at identifying loci that make a small contribution to disease with heterogenous genetic and environmental causes:
diabetes, multiple sclerosis, schizophrenia, bipolar disorder, asthma, heart disease, Parkinson’s disease
Can SNPs explain qunaitiative genetic variation in complex (non-Mendelian) traits?

Hypothesis
Many complex conditions or diseases are caused by alleles of numerous genes
each gene or allele making a small effect to the phenotype
How to identify these genes
Test: how to identify these genes
are any SNP alleles more likely to be associated with a given disease than you would expect if they co-occurred only at random?
Test: selection of SNPs for chip analysis:
Common SNPs→ not rare ones, to maximise variation from limited sample size
International HapMap Project
genotyped millions of SNPs from hundreds of mother-father-child trios from diverse populations to identify ‘haplotypes’

What are haplotypes
genome regions where neiughbouting SNPs show ‘linkage disequalibirium’
i.e non-random association

Why are haplotypes useful
not necessary to genotype every SNP
WHY: although haplotypes are not perfectly maintaine and gradually decay by recombination over many generations
a single SNP from a haplotype still identifies which haplotype is present in many individuals

Result
potential to detect many SNPs that each make a small contribution to disease susceptibility
→ genome-wide association studies (GWAS)

Some maths of association studies
There is a SNP in the angiogenin gene with two alleles, T and G
In an affected sample of 364 patients suffering from ALS (motor neurone disease): G has an allele frequency of 21% (155/728), T has a frequency of 79% (573/728)
In a matched unaffected sample of 299 individuals: G has an allele frequency of 14% (83/598), T has a frequency of 86% (515/598)

Odds Ratio
powerful way to compare the relative risk for ALS conferred by the G allele
Odds Ratio = (155/573 ÷ 83/515) = 1.7

How to test if this association with G with ALS is signficiant?
Chi-squared test:
highly significant P-value <0.001
i.e: the probability that this finding is due to rrandom sampling variation is less than 1 in a thousand

In conclusion:
the association of the G allele with the disease is significant
however→ even this P value needs caution…
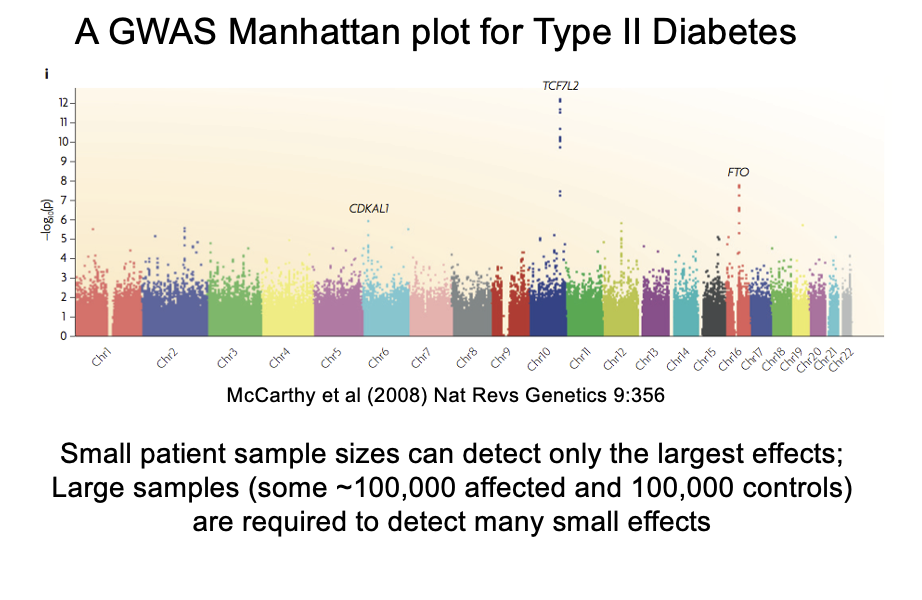
How to interpret association
look at the Manhattan plot
Pitfalls of large-scale SNP analysis
If testing a single SNP→ a P value of <0.001 may appear highly signficiant
but if testing multiple SNPs→ may be an artefact of random variation!
→ with 1M SNPs→ 1000 would show this P-value with random sampling
be scored as false positives (Type I statistical error)
Perhaps angiogenin is causative?→ BUT might just be closely linked to a causative gene?
An artefacts of non-random sampling
e.g a disease more common in particular ethnic groups will show associations with alleles of other loci that are also enriched in the same ethnic group
→ if the control and affected groups are not matched for ethnicity

Caveats
P values used as critieria for statistical significance must be extremely stringent
Even then→never trust an association until it is independently replicated
but how meaningful are strong p values but mall effects anyway?

If an SNP shows irrefutable association with disease→ what does this mean?
Most ‘causative’ SNP are not protein coding→ some affect expression of adjacent genes
Most are of no used for predicition and therapy→ due to low odds ratios
HOWEVER: groups of genes affecting similar pathways/processes offer clues to disease mechanisms
e.g many GWAS hits in multiple sclerosis are at immunity genes
this is only the start of a very long process of biological study
Heritability
genetic contribution to variation
estimated e.g from twin studied
Dark matter genetic contributions on SNP chips? compared to heritbability
genetic contributions of SNPS found in GWAS add up to much less than known heritability
→ There must be some kind of ‘dark matter’ additional genetic contributions that are not SNP chips

What could this dark matter be?
Copy number variants
Omnigenic hypothesis
larger samples can identify ever more genes with ever smaller effects
but: what is the point in knowing that anything can be affected by everything???
Rare alleles of large effect?
Not represented in SNP screens→ may have large effects
recessive disease-causing alleles are now easily identified by exome sequencing

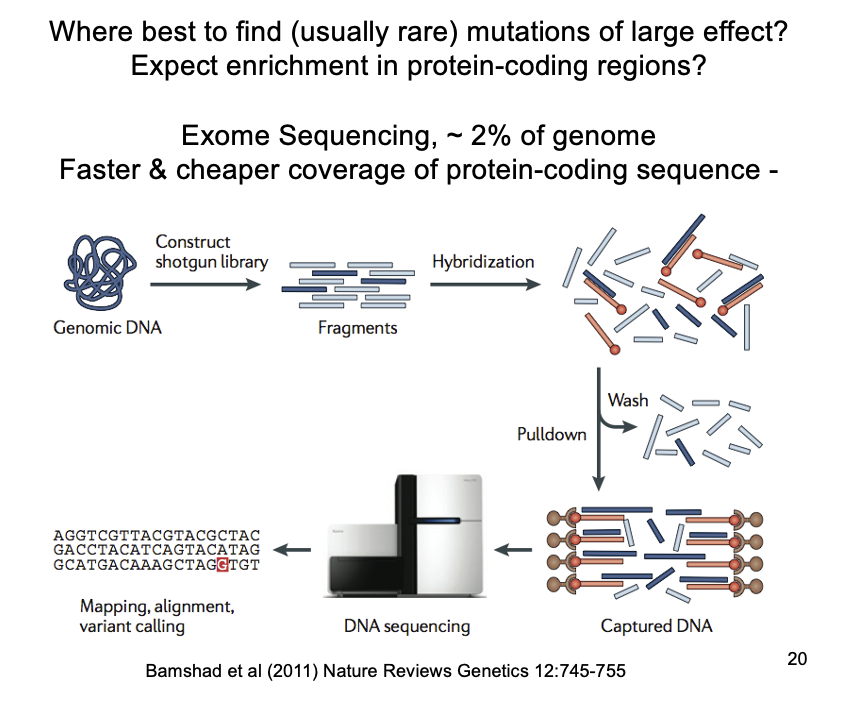
Exome sequencing→ to find rare alleles
exome sequencing of affected individuals and immediate family
exome?→ 2-3% of the genome that codes for exons
BUT CAN”T INTRONS AFFECT GENE EXPRESSION AND DISEASE?
Yields propotionately more severe mutations (affecting coding sequence)→ than the whole genome
for a fraction of the cost and time
NEED: carful interpretation to infer that any mutations identified as causal
Rare recessive alleles – the genetic “dark matter” not detected by GWAS?: how many recessive disease-causing mutations do we carry
Typically (i.e heterozygous) → 150 loss-of-function variants
frameshifts, stops, splice sites disruptions
Most are common enough (>0.5%) to be neutral
BUT→ 10-20 of these are rare variants
i.e likely to be selected against ad pathogenic if homozygous
found in the 1000 genomes project consortium

What do homozygotes for rare disease alleles reveal about disease mechanisms and underlying biology?
→ like screens in model organism
reveal more so than the small effect sizes in brute-force GWAS
depends on humans who are homozygous or ‘compound heterozygous’ (two different alleles in the same individual) for loss of function alleles and understanding their phenotyps
Ever more ‘human knockouts’ are emerging

More human knockouts are emerging via
exome sequencing of patients with persistent undiagnosed disease and immediate families
30-50% of such individuals have good candidate loss-of-function rate alleles for their disease
Phenotypically unbiased sequencing of populations with higher levels of homozygosity:
populations with not many founders or children of consanguineous parents
consanguineous→ relating to or denoting people descended from the same ancestor

Some examples of this
From 2636 Icelandic human genomes, ~8% homozygous loss-of-function for at least one gene
From 3222 Pakistani British exomes, a subset were children of first-cousin marriages; 41% of these were homozygous loss-of-function for at least one gene, most without obvious illness
Screening 874 disease genes in ~500,000 genomes showed 13 individuals homozygous for 8 severe Mendelian conditions, but no symptoms

Human knockouts: other than human knockouts with severse effects as expected…
others are surprisingly mild
any who consent to follow-up studies are a valuable resource for biology and medicine
Example of these mild knockouts
APO3C→ loss-of-function individuals:
have lowered triglycerise lipoproteins in blood
will they be less susceptible to cardiovascular disease?
So is APO3C a plausible therapeutic target?
perhaps genetics may improve on the limited success of big pharma to deliver novel medicines in recent decades???
(rather than incrementally improved)