genomics - dna binding proteins and motif analysis
1/33
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
34 Terms
jacob & monod
repressors encoded in lac operon regulate rate of protein synthesis
protein-dna interaction sequence specificity
-there is no universal code
-electrostatics, hydrogen bonds, water-mediated contacts, and hydrophobic packing
-sequence-specific DNA deformations (indirect readout)
-requires the determinaiton of binding preferences for many members of a family of TFs
Sequence motif
-subsequence with some specific function
-may be in DNA, RNA, protein
-function many be context dependent: ribosome binding site has to be transcribed
-may be gapped or ungapped
Consensus sequence pattern
-may include degenerate bases and allow for mismatches
-search space is over possible patterns
-difficult to obtain an optimal consensus for identifying novel sites
-relative frequency of bases at each positions lost
weight matrix
might go to higher order models
search space is over possible alignments
-more information than a consensus sequence
-many ways to determine the weights
-assumes positional independence
-requires significant data
pattern based algorithms
-can use motif length, num of mismatches, num of seqs,
-4^l patterns, search for most common or significant
N(b,j)
raw scores for a weight matrix model, ie number of times each base showed up in each position
b = base (A, C, T, G), row index
j = jth position in a sequence, column index
F(b,j)
weighted scores for weight matrix model
take raw scores in each position, and get decimal number for likelihood of each base in each position (total should equal 1 in each column)
S(b,j)
probability-normalized log score in weight matrix model
log2[F(b,j)/P(b)]
P(b) = background base distribution
Information content
Sum over columns j and rows b to distinguish divergence of the empirical distribution (f(b,j)) from the background base distribution (p(b))
aka relative entropy, kullback-leibler distance
pseudocounts
entries of 0 in the count matrix cause problems because log(0) is undefined
There’s not enough observations to observe all possibilities
Can add pseudocounts to the matrix to ensure there’s no 0s
protein binding microarrays
can be used for defining PWM
-custom arrays of 60-mer DNA sequences (~44,000 probes)
-contain all possible 10 bp sequences
-each probe contains 27 10-mers
-8-mers guaranteed to occur 16 times
ChIP-seq
-can be used for defining PWM
-cross link protein to DNA
-affinity purify protein-DNA complexes
-reverse cross-links
-identify sequence by hybridization to microarray or by high throughput sequencing
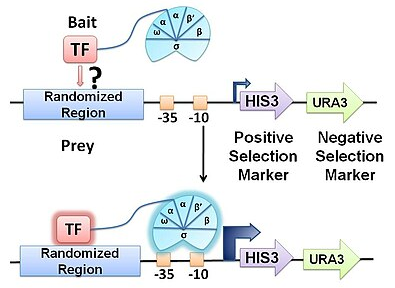
bacterial one-hybrid
method for defining PWM
genetic selection: survival is dependent on DNA-binding
TF of interest is fused to the alpha subunit of RNA pol
randomized library of binding sites created and screened for autoactivation
co transform w TF and selected
library complexity is limited by transformation efficiency
high-throughput SELEX
method for defining PWM
-incubate pure protein with high complexity DNA library
-pull down DNA-protein complexes
-amplify and sequence
JASPAR
open source repository for dna protein binding data
ChIP, PBM, SELEX
>50 different species spanning most clades
extract PWMs for downstream analysis
HOCOMOCO
homo sapiens comprehensive model
public repository of human-specific dna protein data
coverage of nearly all human dna binding domain classes
statistical definition of motif finding
given some sequences, find over-represented substrings (motif discovery)
biological definition of motif finding
given some co-regulated promoters, find transcription factor binding model
class I motif finding algorithms
planted motif problem: single species, multiple genes
-random background sequences
-proper description of a consensus motif gives better models
-randomly plant copies of the motif into sequences
-define an objective function, and use a search algorithm to find the copies that give a good score
exhaustive algorithm
not very tractable
construct every possible combination of alignments and keep the one with the highest information content
given a motif of width w, and k sequences of length l, there are L = (l-w+1) possible locations in each sequence, and L^k alignments to check
greedy algorithm (consensus)
-assume every sequence contains at least one true binding site
-using each l-mer find best match to generate 2-seq alignments
-using top K PWMs to search remaining sequences to include a new sequence
-repeat until all seqs contribute
MEME
Multiple Expectation Maximizations for motif elicitation
-intial “seed” PWM
use the current PWM to determine probability of all positions being sites
reestimate pwm based on the full set of those probabilities
continue until convergence - always convergences to a local maximum
EM is deterministic, meaning it is sensitive to initial seed and may not converge to the global maximum
for this reason, EM should be run multiple times with different seeds
Gibbs sampling
Similar to EM, but some differences:
-initial “seed” pwm
-use the current PWM to determine probability of all positions
-at each iteration, pick one site on each sequence, chosen by its probability to update the PWM, rather than updating using the full set of probabilities
-not guaranteed to converge, but tends to increase objective (IC) and plateau
-can escape local maxima, and therefore not sensitive to seed
gibbs sampling approach to motif discovery
-given “sites”, estimate pattern matrix
-given “matrix”, pick likely sites according to their probability
-iterate between those steps until “convergence”
important: using pseufocounts, and sample sites from estimated prob distribution
how gibbs sampling works
initialization: random assignment of motif locations a1-ak
pick “held-out” sequence
construct initial matrix S from alignment of matrix sequence
score all possible motif locations of held out sequence in A(i,j)
then select a new motif placement randomly based on probability distribution A(i,j)
hold out a new sequence, repeat until matrix converges on better motif window placements
however, sequence may have a placement with no real site, and sequences with more than one site might only have 1 placement
dna binding key take aways
-genome encodes much of its own regulation in protein binding sites
-a full description of the regulatory networks will require identifying these sites
-compact descriptions of the DNA-binding preferences of TFs is afforded by weight matrices
-the information content of an alignment is a measure of specificity
-weight matrix information for a TF is not enough to rule out false positives
-multiple experimental techniques exist for identifying sequences harboring binding sites
-a variety of algorithms can be used to identify motifs in unaligned data
-most predicted binding sites are false positives
class II motif finding algorithms
phylogenetic footprinting
single gene, multiple species
-orthologous background sequences
-sequences linked by a phylogenetic tree
-identify the “best conserved” motif that is under selective pressure
class III motif finding algorithms
multiple genes, multiple species
-combination of phylogenetic data and gene regulation
-use phylogenetic data to reduce search space
-use correlation of motif occurrences among orthologous genes to increase signal strength
CNNs
convolutional neural networks
-sequences are filtered through multiple convolutional layers (based on training sequences) and scored
-filtered sequence scores are pooled and max score retained
-many rounds of convolution → pooling can occur
-fully connected hidden layer used to score sequence
inputs:
-PBM/SELEX
-chromatin accessibility
-ChIP-seq or CUT&RUN/Tag
-principle is to represent sequences that are biologically meaningful in training set
large first filters
represent full motifs
small first filters
learn partial motifs
back propagation
determine the features being learned in early filter layers
-learn discrete sequence contributions to signal
saliency maps
can be used to identify real features that CNN deems important for prediction