BLD 204 Exam 4
1/177
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
178 Terms
neoplasm
a new growth. It is usually abnormal, a mass of tissue, disorderly cells that have disorderly cell proliferation, differentiation and relationship to the surrounding stroma.
-normally there are controls for cell division
-normally cells only divide a certain number of times, only fill so much space and they do this in the right location
normal cell proliferation characteristics
-the cells escape the controls for normal cell division
-they can become basically immortal, dividing many many more times than they should
-the end result is a mass of cells in an inappropriate number in an inappropriate location
disorderly cell proliferation characteristics
-normal cell differentiation is orderly
-it progresses until the cell has reached its normal function and structural characteristics
-tissues are also organized in an orderly fashion
-restriction of the expression of the genome of individual cells allows all of this to happen
characteristics of normal cell division
-neoplastic cells fail to differentiate properly
-they often remain stuck in more immature states
-the more immature the worse the outcome
-immature, undifferentiated cells exhibit behaviors that they shouldn't like expressing fetal proteins or hormones
disorderly differentiation characteristics
-don't invade surrounding tissue
-no metastasis
-growth rate low
-little mitosis
-some atrophy of surrounding tissue by pressure of the mass
biological behavior of a benign neoplasm
-invade surrounding tissue
-metastasis
-lots of mitosis and growth
-damage surrounding tissue
biological behavior of malignant neoplasm
malignancy
invasive, abnormal tissue formations. Increased proliferation and abnormal cell division. Incomplete differentiation where cells vary in size and shape. Nuclei have greater volume than normal in proportion. Abnormal ploidy
carcinoma
malignant tissue/tumor in the epithelium
sarcoma
malignant tumor of connective tissue
dysplasia
cell changes indicative of malignancy but no invasion present yet. It's a warning sign. Common in epithelial tissue.
carcinoma in situ
If the whole depth of the epithelial tissue is dysplastic this is what it is called
hamartoma
tumor like mass that lacks autonomous behavior. Good differentiation and in the right organ. Organization is different from normal. Vascular is the most common
heteroplasia
differentiation of tissue is wrong for the location. Not metaplasia, no change from one fully differentiated form to another. Happens at the stem cell change. Just a mass that doesn't belong there. Masses of various types or one type of tissue.
papilloma
Benign epithelial cells growing in a sheet. Squamous, transitional or columnar.
papilloma and adenoma
benign epithelial neoplasms
adenoma
benign solid islands or masses of cells, arise from the gland or duct epithelium. Because of the source tissue, small groups of cells gather around a lumen but there is no real drainage so a cyst may develop.
G1 -> S -> G2 -> M
cell cycle
G1
phase of cell cycle where cells are not resting like in G0. It can be a very long stage, depends on the cell type. Cells that remain in the cycle are termed being in the growth fraction.
S
phase of cell cycle where DNA synthesis occurs
G2
premitotic phase of cell cycle
M
phase of cell cycle where cells are actively undergoing mitosis
cyclins, cyclin dependent kinases and CDK inhibitors
controls of the cell cycle
hyperplasia
an increase in the number of cells. It is a response to an increase in functional need. Controlled by negative feedback mechanisms
congenital adrenal hyperplasia
hyperplasia with early manifestation. masculization of females, early puberty for males. Salt loss. There are defective adrenal cells which causes decreased cortisol and aldosterone, increased adrenocorticopic hormone from pituitary. Makes more adrenal cells which still doesn't work, built up precursors of hormones get used for steroid production instead
thyroid hyperplasia
hyperplasia caused by an abnormal stimuli or a problem with feedback for TSH. It causes a goiter.
hypertrophy
increase in cell size, it occurs in muscle, the uterus during pregnancy, bladder when the urine outflow is obstructed and heart with increased workload.
atrophy
decrease in bulk due to decrease in demand. Physiological occurs during development especially (ex: in the thymus). Osteoporosis occurs with increased reabsorption, decreased synthesis or both, weight bearing exercises increases demand and helps you lay down bone.
-non neoplastic changes cease when the stimulus is removed
-neoplastic changes don't respond to normal stimuli
-autonomous behavior
difference between neoplastic and non-neoplastic changes
transformation
this refers to cells that have changed into neoplastic cells and exhibit all that behavior. These cells don't show contact inhibition in cell culture.
-normal cells recognize each other, communicate about division, adhere to each other.
-neoplastic cells have a failure of inhibition signals that would restrain proliferation or increased growth factor expression encourages them to proliferate (autocrine, ex: TNF alpha, PDGF) or both things happen
cell communication/proliferation neoplastic vs. normal
-neoplastic cells lose morphology and orientation of mature cells (ANAPLASIA)
-cells that have the most potential to divide in the body are in the stem cell compartment
cell differentiation differences, neoplastic vs. normal
anaplasia
a change in the structure of cells and in their orientation to each other
-DNA to RNA: transcriptional control
-RNA: processing, transport, degradation/stabilization
-RNA to protein: translational control, control of proteins
-environmental control: interactions between cell types, same cell to cell interactions.
controls for daughter cells gene expression
-not fully differentiated
-because they haven't committed to a pathway they can still divide
-the differentiation can be heterogenous
-serious lack of differentiation results in anaplasia
transformed cell differentiation differences
-Self sufficiency of growth signals
-Insensitivity to growth inhibition
-Evading apoptosis
-Limitless replicative potential
-Sustained angiogenesis
-Tissue invasion and metastasis
-reprograming energy metabolism
-evading the immune system
hallmarks of cancer
most growth factor action on normal cells is paracrine. Cancer cells make their own or they tell the stroma to make them some.
cancer cells growth factors (self sufficiency of growth signals)
cancer cells overexpress these receptors or they have mutated receptors that are "always on" sending signals continuously even without growth factors. Examples of this is overexpression of EGF (occurs in squamous cell carcinomas of lung, glioblastomas and epithelial tumors of head/neck) and similar overexpression of ERBB2 (happens in some breast cancers)
cancer cells and growth factor receptors (self sufficiency of growth signals)
mutations occur further downstream, on the signally proteins inside the cell. Direct stimulation of signal to the nucleus without growth receptor signal. Two most common mutations are in RAS and ABL.
Cancer cells and signal tranducing proteins (self sufficiency of growth signals)
RAS
signal tranducing protein, most commonly mutated gene in human tumors. Small protein that binds GTP and GDP
ABL
tyrosine kinase. Translocation in chronic myelogenous leukemia from chromosome 9 to 22. fuses with BCR (breakpoint cluster region) there and makes fusion protein that has constitutive activity
cancer related mutations in transcription factors can result in expression of more growth promoting genes like growth factor genes. Ex: MYC
nuclear transcription factors and cancer cells (self sufficiency of growth signals)
MYC
nuclear transcription factor that activates or represses. It activates genes that encourage progression through the cell cycle like cyclins. It represses genes that repress genes that prevent it. Encourages aerobic glycolysis and glutamine utilization. Mutation of it is seen in Burkitt's lymphoma
cyclins
regulate progression through the cell cycle
cyclin dependent kinases
activated by binding to cyclins, maintain orderly progression
cyclin-dependent kinase inhibitors
broad and selective inhibitors of CDKS
-growth factors
-growth factor receptors
-signal tranducing proteins
-nuclear transcription factors
-cyclins and cyclin dependent kinases
what factors play into the self sufficiency of growth signals hallmark of cancer?
-all cancers seem to have mutations that disable the G1-S checkpoint
-increases cyclin D and CDK4 expression
-CDKIs disabled by mutation or silenced
cyclins and cyclin dependent kinases and cancer cells (self sufficiency of growth signals)
tumor supressor genes
brakes for proliferation. The result of mutations in this system is very similar to self sufficiency of growth signals
-RB (retinoblastoma protein)
-TP53 (tumor protein 53)
-TGF beta (transforming growth factor beta)
-contact inhibition pathways
four major gene targets of insensitivity to growth inhibition signals
RB
DNA binding, regulates the G1-S checkpoint. Important in development, cell cycle "clock" once past, they must divide eventually. Fairly common mutation. Many viruses which are oncogenic bind this (ex: HPV). This is the gene implicated in retinoblastoma which is a rare childhood cancer
TP53
monitors cell stress. Anoxia, DNA damage, inappropriate signaling. Encourages transcription of CDKI which inhibits the cell cycle. Blocks G1 to S for a time for repairs. If no repair, induces senescence or apoptosis. Viruses can subvert it just like RB (ex: HPV)
TGF beta pathway
normal proliferation inhibitor in epithelial, endothelial and hematopoietic cells. Mutations in 100% of pancreatic cancers and 83% of colon cancers. This is a loss of function of inhibitor
contact inhibition
cadherins mediate cell to cell contact. NF2 gene product merlin facilitates E cadherin contact inhibition. APC gene product loss also can be involved in loss of this by destroying beta catenin which can be a growth promoting transcription factor.
TCF is continually activated
loss of function of APC leads to cell proliferation because...
BAX-BAK pro apoptotic action is regulated by BCL2 which is anti apoptotic. BCL2 mutations that activate it are common in B cell lymphomas. When BCL2 is always activated (like in cancer) apoptosis won't occur. Cancer cells also avoid autophagy by mutation or take it over to get parts to grow.
evading apoptosis
60-70
max number of cell divisions is usually...
Short telomeres are recognized as dsDNA breaks leading to senescence mediated by RB and p53
how does senescence occur?
RB and p53 are inactivated and the ends of the two chromosomes get connected in a last ditch effort at repair, this leads to dicentric chromosomes which lead to mutations.
limitless replicative potential (the big picture)
telomerase
usually only present in stem cells. It maintains telomere length. It can be reactivated to maintain telomere length in cancer cells (85-95%)
-constant proliferation leads to telomere shortening
-telomere shortening leads to bridge fusion breakage cycle when p53 is absent
-BFB cycle leads to mutation
-telomerase is reactivated, fixing mutations and allows the cells to continue dividing
limitless replicative potential model
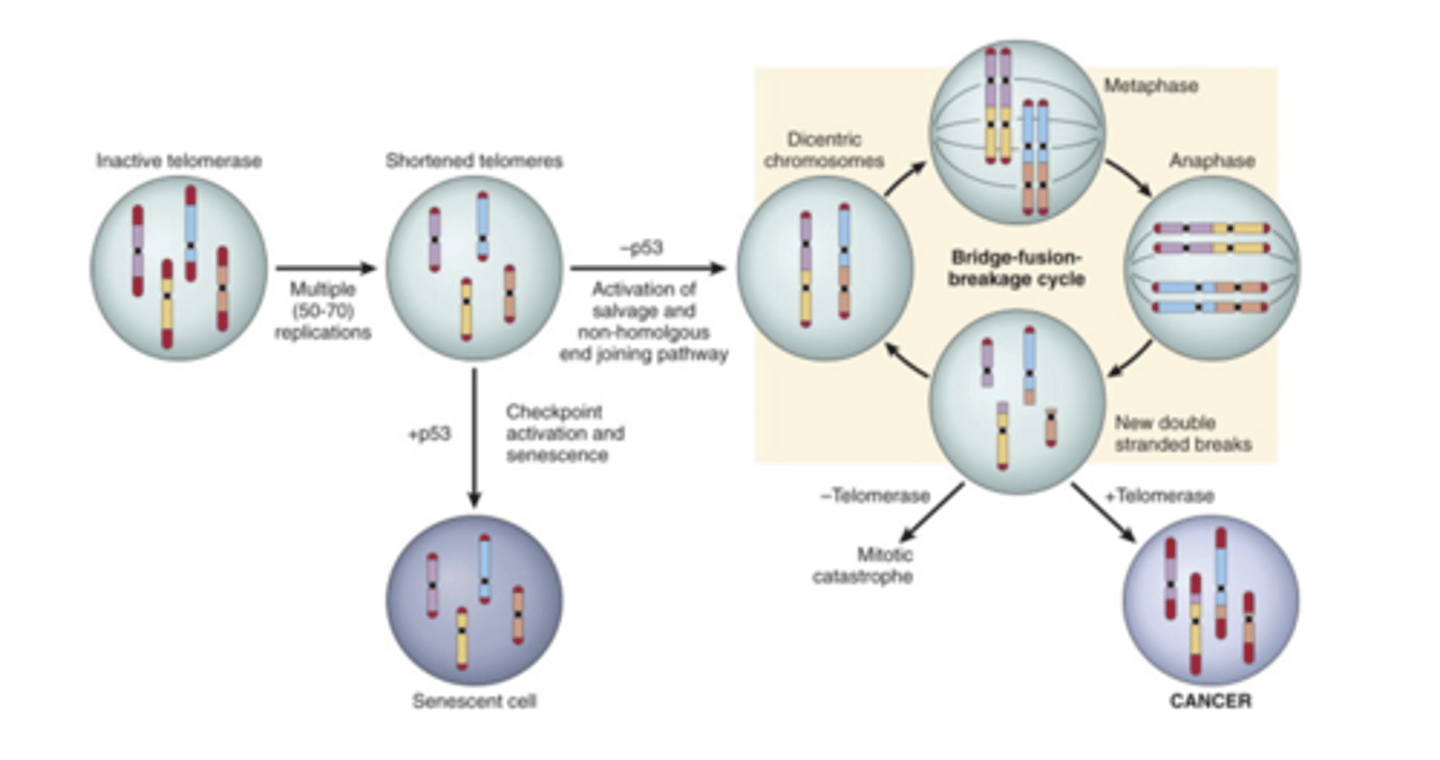
Vascularization
tumor bigger than a 2mm can't survive without...
-neoangiogenesis: new sprouts from capillaries in the area
-vasculogenesis: endothelial cells come from the bone marrow to make blood vessels
how are tumors vascularized?
hypoxia stimulates productions of pro-angiogenesis cytokines like VEGF which happens through the activation of HIF-1 alpha. Unless there is hypoxia, HIF-1 alpha is controlled by VHL which binds its signaling for destruction. If there is hypoxia, VHL no longer binds HIF-1 alpha.
development of sustained angiogenesis: hypoxia
Thrombospondin-1 (TSP-1) is produced by stomal fibroblasts and synthesis is induced by p53. Protease produced angiostatin, endostatin and vasculostatin
development of sustained angiogenesis: inhibitory factors
loss of p53 causes loss of TSP 1, VHL mutations are associated with renal cancers, cancer cells produce VEGF and proteases that can swing the balance.
development of sustained angiogenesis in cancer
metastasis
second tumor site with no continuity with the first. Spread via the lymphatics or the blood stream
most common in carcinomas (ex: breast or malignant melanoma). Invades the lymphatic vessels and spreads up along the lymphatic vessel to the node. The blood stream and lymphatics are connected
lymphatic spread (ability to invade and metastasize)
most common in sarcomas. Invasion can happen in the tumor's new blood vessels or in nearby vasculature
spread via bloodstream (ability to invade and metastasize)
1. liberation
2. invasion
3. transfer as emboli
4. adhesion at endothelium
5. migration from the vessel
6. survival (angiogenesis)
7. multiplication and growth
steps of metastasis
GI and pancreas, spread via blood vessels
most common source of liver cancer
-liver
-skeleton
-brain
-lung
-adrenals
common secondary sites for metastasis
breast, prostate, kidney, thyroid
common sources of skeleton cancer
lung is most common, also breast and adrenals
common source of brain cancer
breast and stomach carcinomas and sarcomas
common source of lung cancer
lung and breast
common source of adrenal cancer
loss, gain
(gain/loss) of p53 and (gain/loss) of telomerase causes unlimited replicative potential
-anaerobic glycolysis (Warburg Effect)
-Favored when rapid growth is required
reprograming energy metabolism
-most cancer patients are immunocompetent until treatments
evading the immune system
-growth promoting proto oncogenes
-growth inhibiting tumor suppressor genes
-genes that regulate apoptosis
-genes that are involved in DNA repair
four classes of regulatory genes that are targets of genetic damage
oncogenes
genes that include a transformed phenotype when expressed in cells. Usually mutated or overexpressed versions of normal genes proto oncogenes. DOMINANT: single allele is sufficient for transformation
tumor supressor genes
genes that normally suppress uncontrolled growth. When they are mutated or lost, the transformed phenotype appears. Usually, both alleles have to be damaged for the phenotype to appear but sometimes a single allele will do it (haploinsuffiency). There are two types, governors and guardians
governors
type of tumor suppressor gene where a mutation removes an important stop mechanism (RB)
guardians
type of tumor suppressor gene that produces proteins that act as sensors of genomic damage (p53)
BCL2
an example of a proto oncogene that regulates apoptosis is...
karyotype changes
changes in the number and appearance of chromosomes. Specific changes have been identified with particular neoplasms
-karyotype changes
-balanced translocations
-deletions
-gene amplifications
-aneuploidy
-microRNAs and cancer
-epigenetics
-selective pressures
genetic lesions that lead to cancer
balanced translocations
translocation moves the gene to where its under an inappropriate, highly active protomer. Translocations make fusion proteins. Lymphocytes and their precursors are the most common types of cells where genome rearrangements occur. This is because these cells intentionally make DNA breaks in rearranging their antibody or TCR genes. This occurs in MYC and Burkitt's lymphoma and follicular B cell lymphoma and BCL2
deletions
second most common type of karyotypic abnormality. Large ones are more common in hematopoietic solid tumors. Often this occurs to tumor suppressors. Causes loss of heterozygosity, point mutation in one allele and deletion of the other.
amplifications
Extra copies of particular genes. This occurs in NMYC neuroblastoma and ERB2 breast cancer
aneuploidy
number of chromosomes that is not a multiple of normal haploid number (23). This occurs because of mitotic checkpoint errors.
MicroRNAs and Cancer
non-coding single stranded regulatory RNA. Oncogene expression is increased and tumor suppressor expression is decreased.
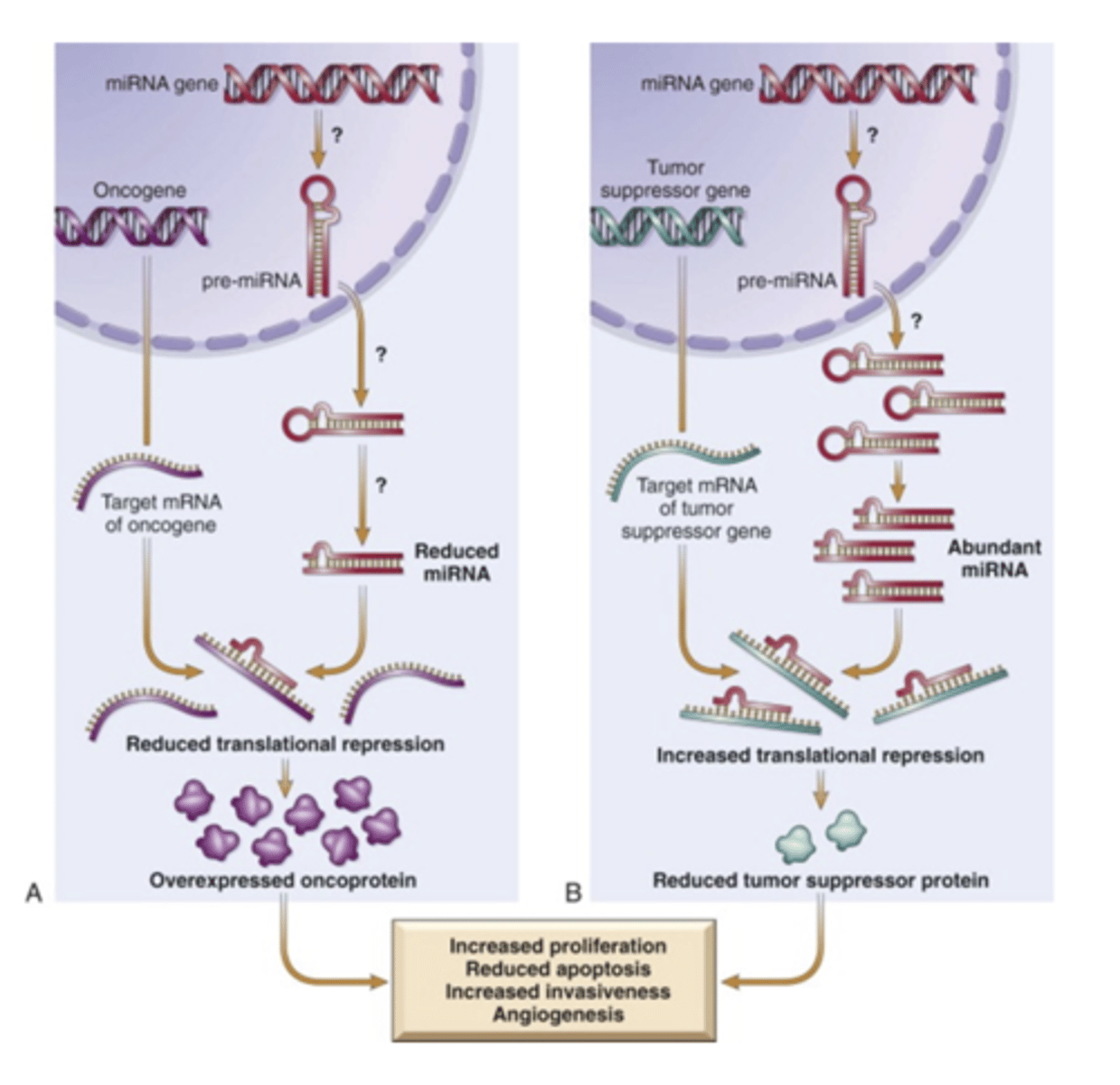
epigenetics
reversible, heritable changes in gene expression. This is not a mutation. Methylation of histones and DNA change expression of genes. Cancer cells have global hypomethylation of the genome and hypermethylation of certain protomers.
hypermethylation
epigenetic change that silences gene expression. Promoters of tumor suppressor genes
hypomethylation
epigenetic change that makes the genome unstable and leads to tumors in mice
selective pressures
multiple genetic alterations which result in transformed phenotype. Tumor progression, the fact that cancer becomes more malignant with time is a phenomenon that has been observed often. Initial tumor is monoclonal, but by the time cancer reaches its dangerous stage clinically, they may be very heterozygous because the tumor cells are undergoing darwinian selection. This can be immune and non-immune. Subclones that bear advantageous mutations out compete those that don't. Tumors that recur are more aggressive and more resistant to treatment
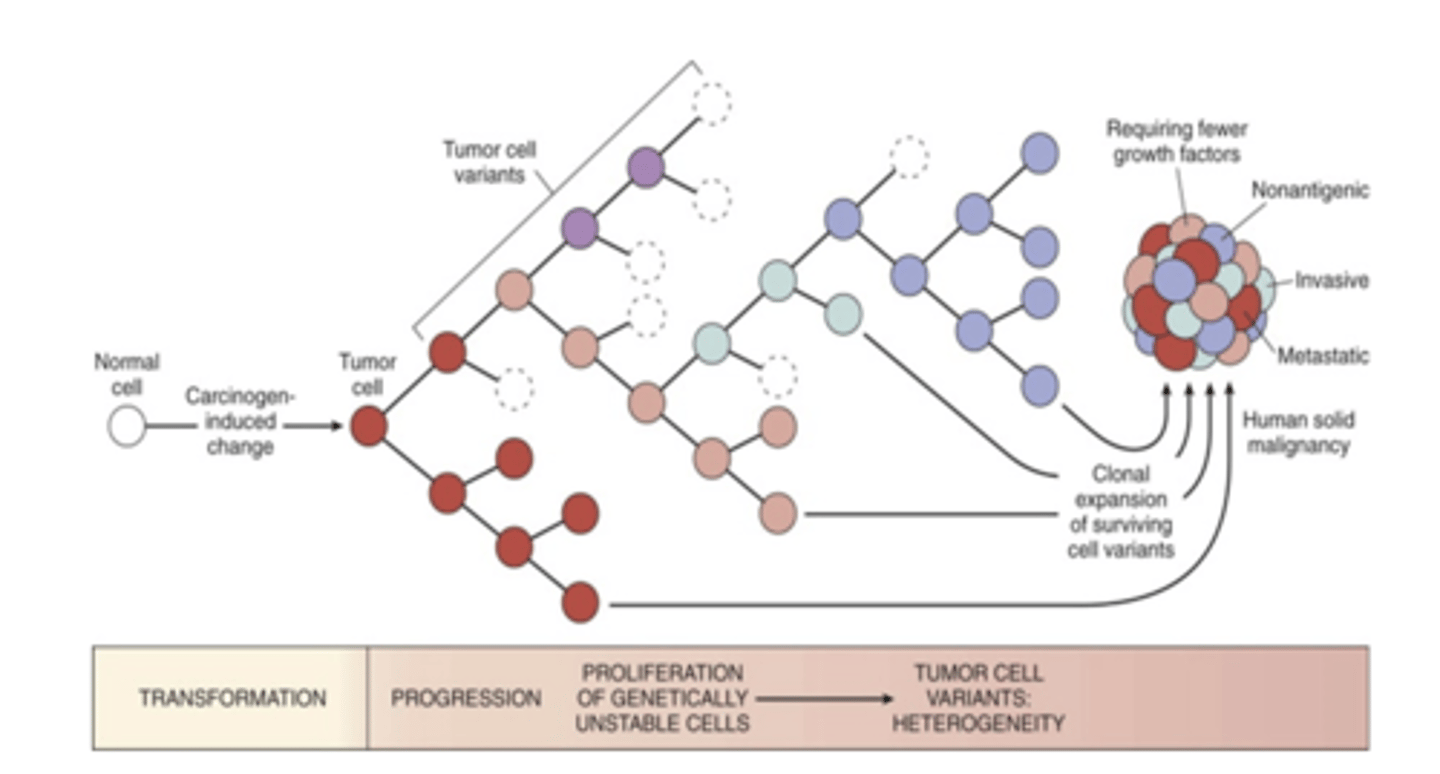
-mismatch repair
-nucleotide excision repair
-recombination repair
the three repair systems
Hereditary Nonpolyposis Colon Cancer Syndrome
problems with mismatch repair. Carcinoma of the cecum and proximal colon. Mismatch repair genes proofread DNA to make sure mismatches don't accumulate. At lease four genes are implicated. The effect is indirect, it allows mutations in genes that contribute to the hallmarks.
Xeroderma pigmentosum
mutations contribute to higher risk of sun damage from UV light. UV cross linking of pyrimidine is not repaired by the excision process (problem with nucleotide excision). Several proteins are involved, loss of one is enough to cause a problem
Fanconi anemia
problem with recombination repair. Group of autosomal recessive disorders. Phenotypes are complex and characterized by other issues too, like anemia. Also BRCA1 and BRCA2 in breast cancer, chromosomal breaks and severe aneuploidy.