Bacteria and Yeasts
1/48
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
49 Terms
Advantages of working with bacteria.
Simple structure (no nucleus)
Good genetic model, e.g. E. coli genome has been fully sequenced and studied
Good experimental model: bacterial cells are easy to manipulate and culture in minimal nutrition environments.
Short generation time
Non-pathogenic: certain E. coli strains are harmless, they are part of the normal flora of the human digestive tract.
Can host plasmid DNA
Approximate number of genes in E. coli
5000
Prokaryotic models gave basic insight in… (10 bullet points)
Mobile genomic elements (transposons)
Horizontal gene transfer (conjugation, transformation, and transfection)
DNA replication, transcription, and translation
Gene/protein function
Principles of transcription regulation (e.g. Lac operon)
Signal transduction mechanisms (e.g. two component systems)
Metabolic pathways (e.g. respiration) and networks
Tools for recombinant technology
Evolution
Cellular differentiation
And many more …
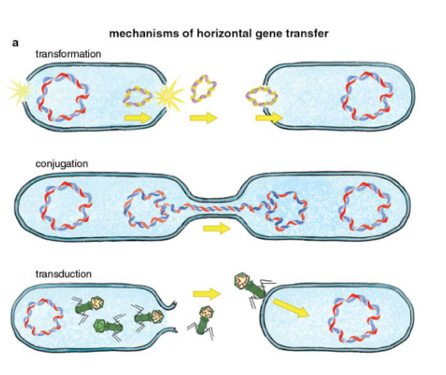
Lac Operon
Lactose and glucose present
Presence of lactose means lac repressor is inactivated and cannot bind to lac promoter --> transcription possible.
However, presence of glucose means no cAMP is produced --> no cAMP-CAP complex --> low transcription.
Lactose absent, glucose present
Absence of lactose means lac repressor is activated and represses the lac operon --> transcription not possible regardless of glucose concentration.
Lactose present, glucose absent
Presence of lactose means lac repressor is inactivated and cannot bind to lac promoter --> transcription possible.
Absence of glucose means cAMP-CAP complex can activate the lac operon --> high transcription.
Lactose and glucose absent
Absence of lactose means lac repressor is activated and represses the lac operon --> transcription not possible regardless of glucose concentration.
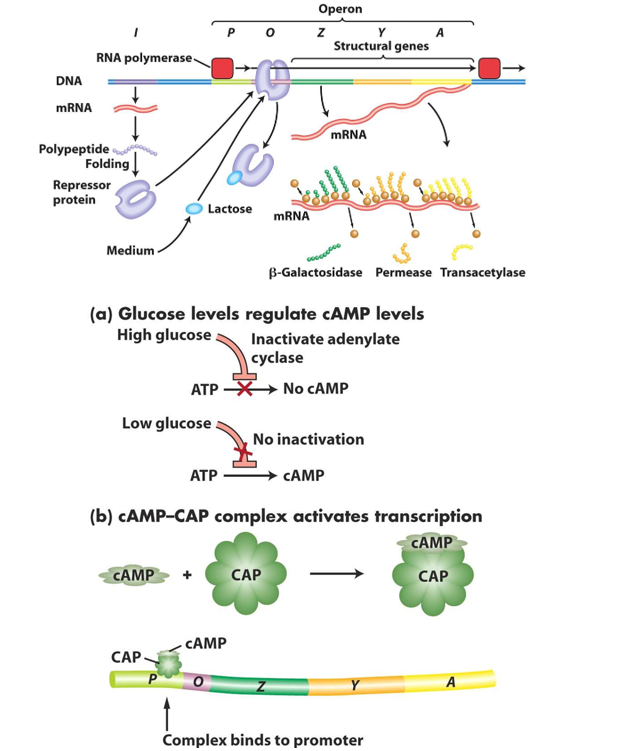
Briefly explain what two component systems are in bacteria.
Two-component systems (TCS) in bacteria are signal transduction mechanisms that help them sense and respond to environmental changes.
The main type of two-component systems are the Histidine Aspartate Phosphorelay (HAP) systems. They consist of two main components:
histidine protein kinase (HPK): a membrane bound sensor proteins that detect environmental stimuli (e.g. pH, osmolarity, the presence of specific molecules). Upon activation, it auto phosphorylates at a conserved histidine residue.
aspartate response regulator (RR): a cytoplasmic protein that receives the phosphate group from the histidine kinase, usually at an aspartate residue. This phosphorylation alters the response regulator’s activity, leading to changes in gene expression for example.
The figure shows examples of HAP systems
The majority of the genetic information on an individual comes from …
microbes.
Slime mold
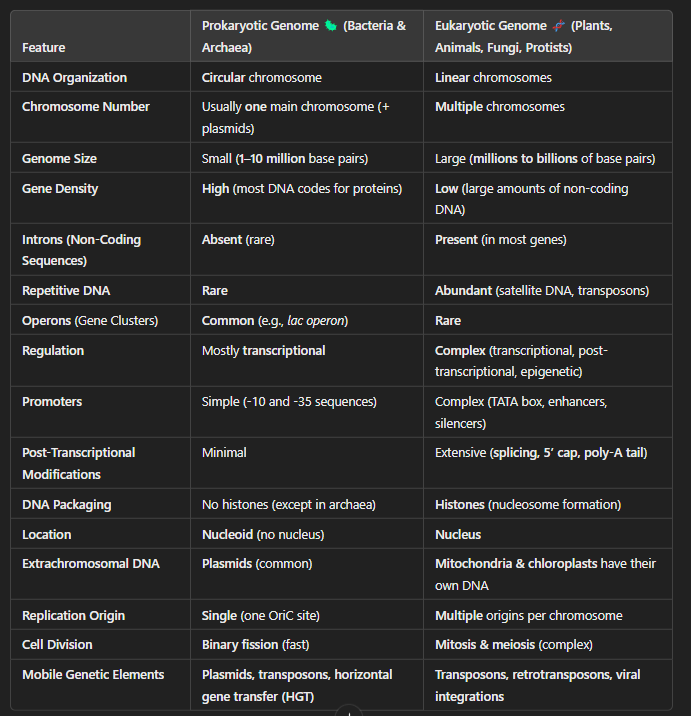
Prokaryotic vs Eukaryotic Genome
Open Reading Frame (ORF)
An open reading frame (ORF) is a continuous sequence of codons in a DNA or RNA molecule that has the potential to be translated into a protein. It begins with a start codon (AUG in RNA) and extends until it reaches a stop codon (UAA, UAG, or UGA in RNA).
Alternative splicing gives information about …
which parts of the protein are functionally important.
For example, if in each alternative splicing, the same exons end up in the final mRNA, we know that this part of the protein is functionally important. The exons that do not always end up in the final mRNA product are not essential.
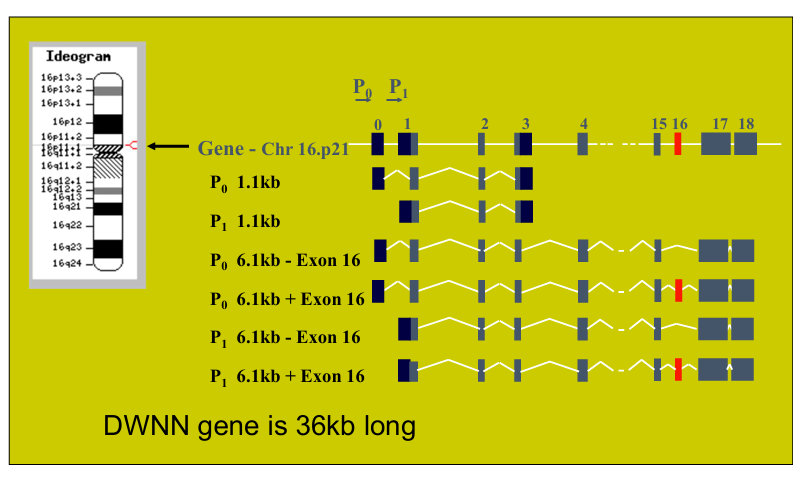
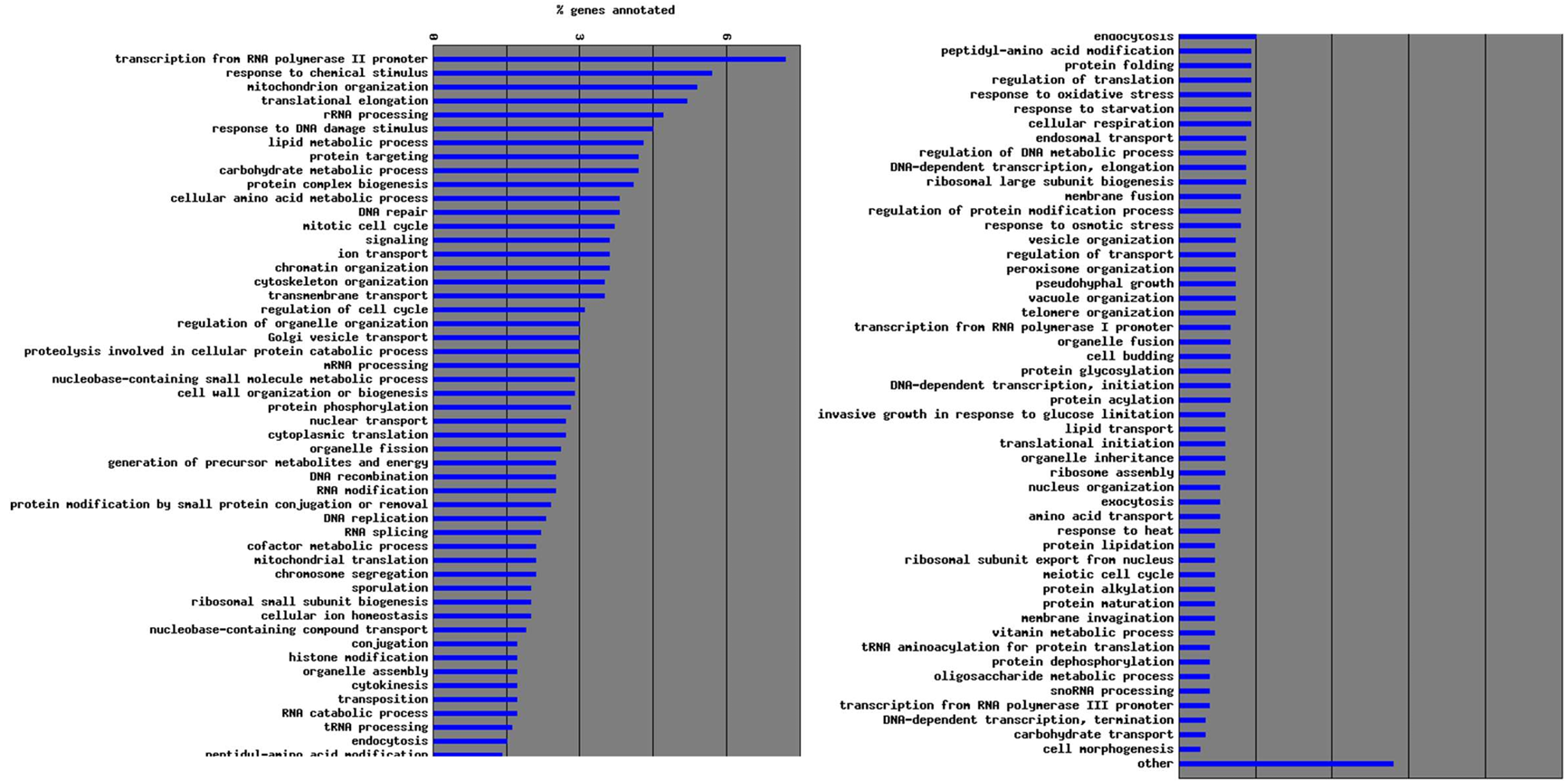
Compare Saccharomyces Cerevisiae and Schizosaccharomyces Pombe.
S. pombe is the fission yeast, while S. cerevisiae is the budding yeast. As the names indicate, S. pombe reproduces through binary fission (like humans), while S. cerevisiae reproduces through budding (asymmetric division)
Their genomes are similar in size:
13.8 Mb with 5070 ORFs for S. pombe
12.1 Mb with 5600 ORFs for S. cerevisiae
Despite their similar sized genomes, they are neither related nor syntenic (genes or genomic regions do not maintain the same relative order or location on chromosomes).
Introns: S. pombe has more introns (~5000) than S. cerevisiae (~250).
Number of chromosomes: S. pombe has 3 chromosomes while S. cerevisiae has 16.
Centromeres: The centromeres of S. pombe are large, complex, and repetitive. They are more similar to mammalian centromeres compared to those of S. cerevisiae, which are small.
Genes: S. pombe shares some genes with humans that are missing from S. cerevisiae. Thus, fission yeast is a complementary experimental system to budding yeast.
S. Pombe better model than S. Cerevisiae for certain processes: For example, S. Pombe has:
an RNAi pathway
repetitive centromeres
G2/M cell cycle control
complex heterochromatin and splicing regulation
What makes saccharomyces cerevisiae a good model organism? (11 bullet points)
Small, well characterised genome (first eukaryotic organism to have its entire genome sequenced).
Unicellular eukaryote: it shares many fundamental processes with human and other eukaryotes, but the fact that it is unicellular greatly simplifies its study.
Correspondence with human genes (approx. 30% conservation with human disease genes).
Ease of genetic manipulation: homologous recombination is highly efficient, allowing to introduce mutations in the genome.
Transformation is efficient (although not as efficient as in E. coli).
Relatively simple gene structure (few introns, promoter regions and regulatory elements well understood).
Rapid growth rate: doubling time of 90 to 120 minutes, allowing for high-throughput experiments.
Cheap
Easy to handle compared to other eukaryotes like mammalian cells.
Can exist both in diploid and haploid form: this gives researchers more flexibility in genetic analysis, especially when studying recessive mutations, mating, meiosis, and recombination.
They can ferment (anaerobic, doesn’t require mitochondria) or respire (aerobic, requires mitochondria): this means you can make mutants in mitochondrial genes and still grow them under fermentative conditions. You can also shift between conditions to test the impact of the mutations.
S. cerevisiae VS Human chromosomes

In S. cerevisiae, the intergenic space is between 200 and 1000 bp, making the yeast genome …
highly compact (about 1 gene per 2 kb).
Gene Ontology
Gene ontology is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
The projects aims to:
Maintain and develop a controlled vocabulary of gene and gene product attributes.
Annotate genes and gene products, and assimilate and disseminate annotation data.
Provide tools for easy access to all aspects of the data provided by the project.
Three main attributes: molecular function, biological process, and cellular component.
In GO, what does the cellular component attribute represent?
Represents where the gene product of interest is located.
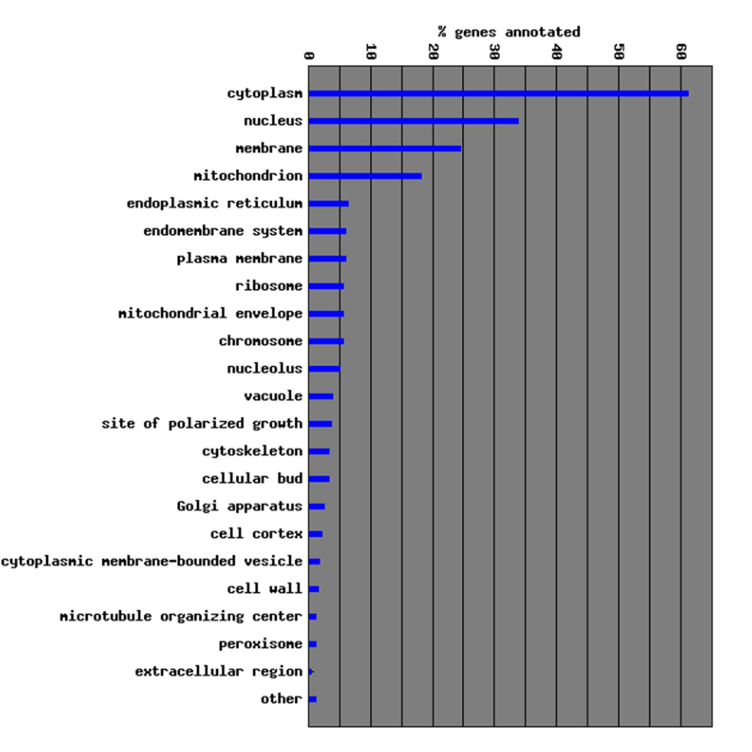
In GO, what does the biological process attribute represent?
Represents in which biological process(es) the gene product of interest is involved in.
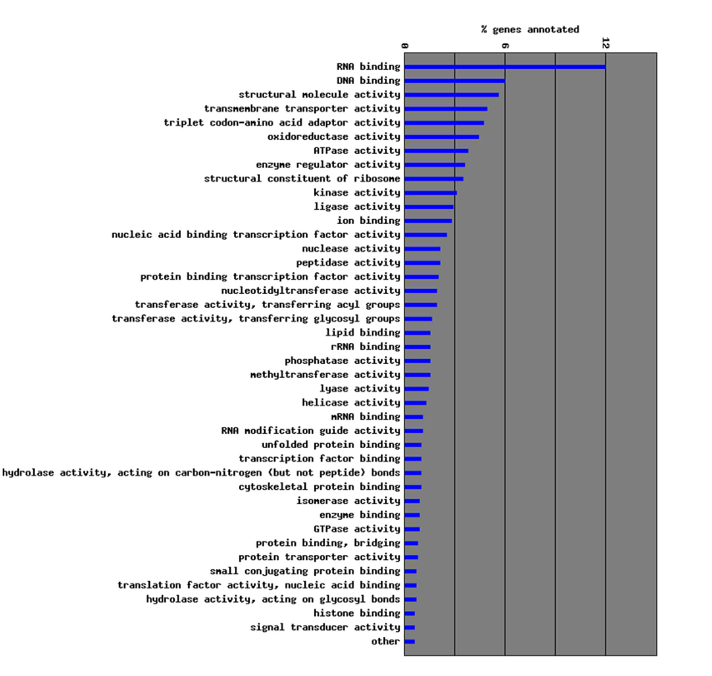
In GO, what does the molecular function attribute represent?
Yeast gene nomenclature
Gene names consist of 3 letters and up to 3 numbers (sometimes meaningful)
Wild type genes are written with capital letters and in italics (e.g. TPS1, RHO1).
Recessive mutant genes are written with small letter in italic (e.g. tps1, rho1).
Mutant alleles are designated with a dash and a number (e.g. tps1-1, rho1-23).
Mutations that have been constructed by gene deletion are indicated with a capital delta (Δ). The genetic marker used for deletion is also indicated (e.g. tps1Δ::HIS3 means that the HIS3 gene, i.e. the genetic marker, was inserted in place of tps1).
The gene product (protein) is written with a capital letter at the beginning and not in italic. A p is often added at the end (e.g. Tps1p, Rho1p).
Genes whose function has not yet been determined get a landmark name:
The first letter is Y for yeast
The second letter represents the chromosome (A = I, B = II, C = III …).
The third letter corresponds to whether the gene is on the left or right arm of the chromosome (L or R).
The three digit number stands for the ORF counted from the centromere on that chromosome arm.
The last letter indicates if the strand is Crick or Watson (C or W).
Some genes do not follow this nomenclature (e.g. HO, MATa …).
What are the advantages of yeast being able to exist as a diploid and haploid organism? (4 main points)
It gives researchers more flexibility in genetic analysis, especially when studying recessive mutations, mating, meiosis, and recombination.
Studying recessive mutations
In the haploid state, every gene has only one copy, so mutant phenotypes are directly observable, even if they are recessive.
Example: we can easily figure out if a gene is essential by knocking out the gene, and observing if the cell survives or not.
Genetic complementation and interaction studies
In the diploid state, you can complement mutations by providing a wild-type copy of a gene to see if it rescues the mutant phenotype.
You can also study dominance relationships between alleles and interactions between different mutations (like synthetic lethality).
Mating and meiosis
Haploid yeast of opposite mating types (a and α) can fuse to form a diploid, which allows controlled crosses and strain construction.
Diploids can undergo meiosis and sporulation, producing haploid spores, perfect for dissecting genetic traits across generations.
Switching between haploid and diploid
Yeast can switch between haploid and diploid states naturally or in the lab, which means you can design experiments to create mutants in haploids, mate them into diploids, and analyse progeny from meiosis.
Explain S. cerevisiae cycle
Functional analysis tools from yeast
Genome-wide deletion collections: library of mutant strains, where each strain has a specific gene deleted (knocked out). Each strain usually has a selectable marker (like HIS3, KanMX) inserted in place of the gene, as well as molecular barcodes, so you can track them in pooled experiments.
Genome-wide overexpression collections: library of plasmid or strains where each gene in the genome is cloned under a strong inducible promoter so that the gene is overexpressed. Each strain overproduces one specific protein when induced.
Genome-wide tagged collections: library of yeast strains where each gene is tagged with a detectable marker. This helps study where certain proteins are expressed, in what quantity, their interactions, and their functions.
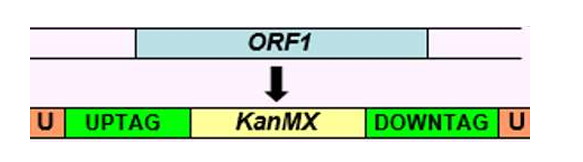
How are DNA fragments for gene knockouts generated?
To knock out a gene, you need to replace it with a marker, and the yeast must be able to recognize the DNA as homologous and swap it in.
This is done by flanking the marker with regions that match the sequences upstream and downstream of the target gene (i.e. knocked out gene).
How is this done?
Technique 1
The upstream and downstream flanking sequences of the genes are PCR amplified.
They are then fused to a selectable marker (e.g. URA3) in a second PCR reaction.
The primers used to amplify the marker must have extra sequences at their 5’ ends that match the flanking regions of the gene you are deleting.
Technique 2
It can also be done in one PCR reaction, where only the marker is amplified.
Recombination is mediated by the primer sequences.
Techniques to study protein-protein interactions.
Yeast-2-hybrid (Y2H)
Split GFP/BiFC (Biomolecular Fluorescence Complementation)
FRET
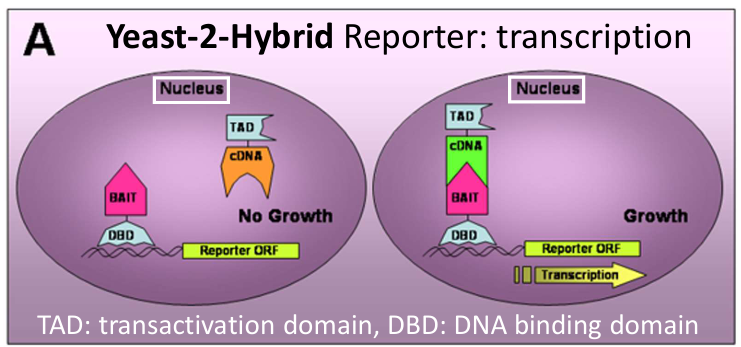
Explain the yeast-2-hybrid method.
What?
The Yeast Two-Hybrid (Y2H) system is a method used to detect protein–protein interactions in vivo, specifically within the nucleus of yeast cells.
How?
The Y2H system is based on modular transcription factors which have:
A DNA-binding domain (DBD): binds to a specific DNA sequence.
A transcription activation domain (TAD): recruits the transcriptional machinery.
On their own, neither domain can activate transcription, they must be together on the same protein (or brought into proximity) to initiate transcription of a reporter gene.
The protein of interest (bait) is fused to a DBD, and a potential interacting partner (prey) is fused to a TAD. Only if the two proteins interact will the DBD and TAD be brought together and induce transcription. This leads to transcription of the reporter gene.
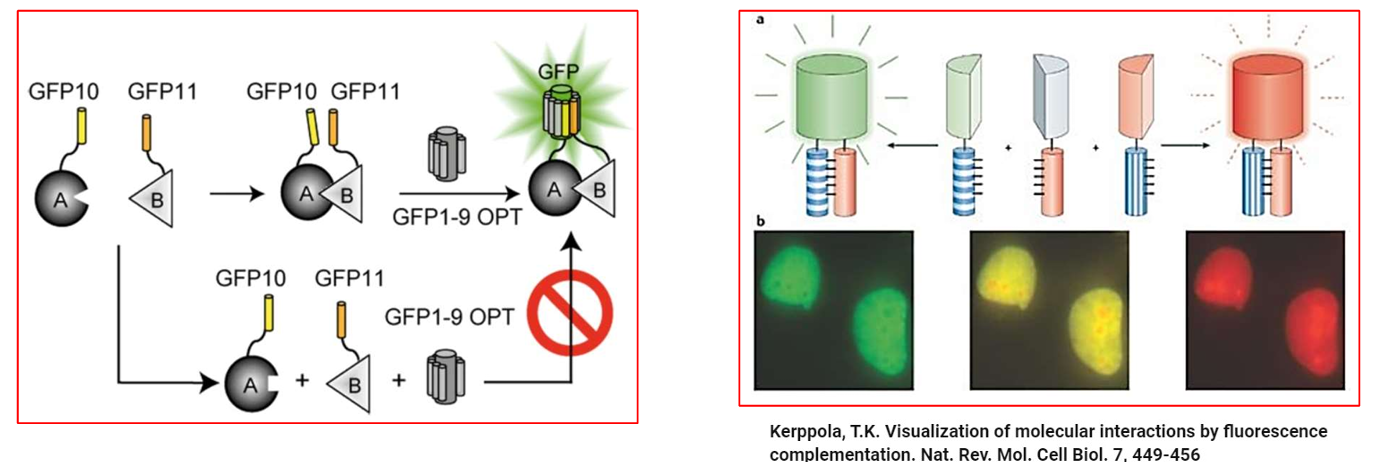
Explain split GFP/BiFC (Biomolecular Fluorescence Complementation).
A fluorescent protein like GFP can be split into two non-fluorescent fragments: e.g., GFP10 + GFP11.
These fragments are fused to two proteins of interest (A and B).
If proteins A and B interact, the GFP fragments come together → reconstitute a functional GFP → fluorescence is observed.
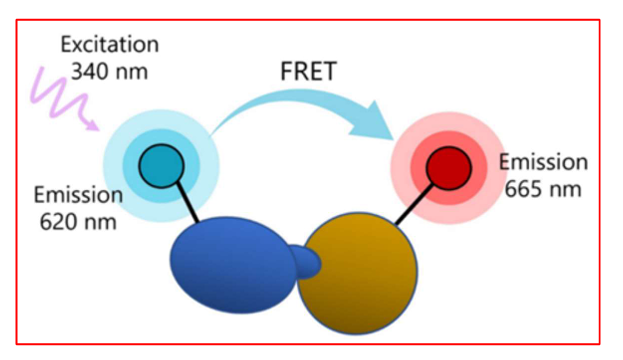
Explain how FRET is used to study protein protein interactions.
Two proteins are tagged with different fluorophores:
Donor (e.g., CFP) emits light upon excitation.
Acceptor (e.g., YFP or RFP) only emits light if it’s very close to the donor (~1–10 nm).
If the tagged proteins interact or come very close, energy is transferred from donor → acceptor → emission from the acceptor is detected.
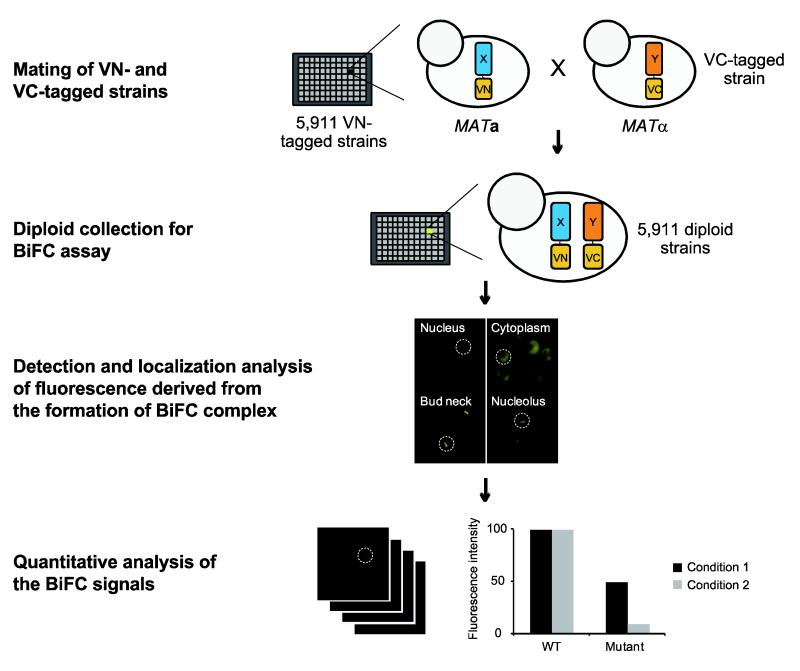
How can BiFC be scaled up to a genome-wide screen in yeast?
BiFC (Bimolecular Fluorescence Complementation) can be used at a genome-wide scale to detect protein–protein interactions in yeast.
VN: N-terminus of the Venus fluorophore
VC: C-terminus of the Venus fluorophore
Mating of VN- and Vc-tagged strains: A yeast strain (MATalpha) expressing a VC tagged protein is mated with a library of 5911 strains (MATa) each expressing a different VN-tagged protein. These proteins are potential partners of the VC-tagged protein.
Diploid collection for BiFC assay: each diploid strain co-expresses one VN-tagged protein and one VC-tagged protein. If the two proteins interact, the split fluorescent protein fragments come together and reconstitute fluorescence.
Detection and localisation analysis: cells from each diploid stains are examined under a microscope. Fluorescence means that the tagged protein of that strain are interacting. Subcellular localisation of fluorescence indicates where this interaction occurs.
Quantitative analysis: intensity of fluorescence is measured to quantify interaction strength.
This allows researchers to not only identify interactions, but also visualize where they occur in the cell and quantify their strength under different conditions.
Techniques to generate genetic interaction networks
Epistasis analysis: for determining gene pathways and how they interact.
Complementation assays: tests whether two mutations causing a similar phenotype are in the same gene (i.e. different alleles of the same gene) or in different genes
Synthetic genetic arrays (SGA): to systematically investigate genetic interactions between a query gene and a library of mutants (e.g. deletion, overexpression…)
Enrichment screenings: to identify mutants that grow better under a selective condition.
Chemogenetic screening: to map drug effects to specific genes or pathways.
What is epistasis analysis?
Epistasis refers to a genetic interaction where the effect of one gene (locus) is masked or modified by another gene. This is useful for determining genetic pathways and the order of gene function.
🔹 Purpose:
To determine the order of genes in a biological pathway and how they interact.
🔹 Method:
Create double mutants for two genes (e.g., gene A and gene B).
Observe the phenotype of the double mutant.
Compare it to the single mutants:
If the phenotype of the double mutant resembles one of the single mutants, the gene with that phenotype is epistatic (i.e., downstream or dominant in the pathway).
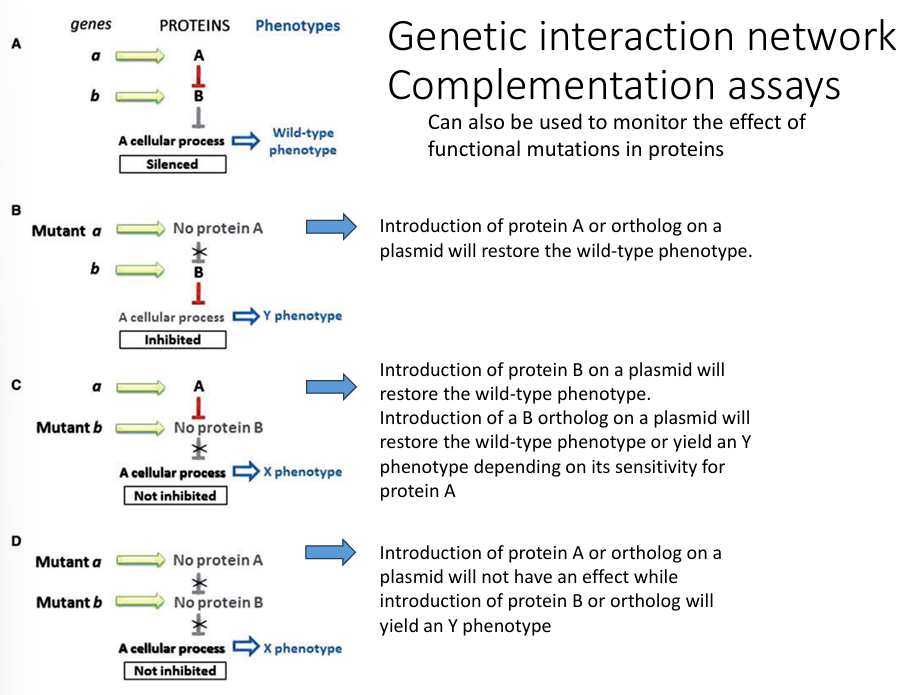
What is a complementation assay?
Complementation tests determine whether two mutations causing a similar phenotype are in the same gene (allelic) or in different genes.
🔹 Purpose:
To identify whether mutations affect the same gene or different genes.
🔹 Method:
Cross or co-express two mutants with similar phenotypes (e.g., two strains of yeast with white colonies instead of red).
Observe the phenotype of the heterozygote or diploid.
Interpret:
If the phenotype is rescued (wild-type) → mutations are in different genes → complementation occurred.
If the phenotype is still mutant → mutations are in the same gene → no complementation.
Synthetic lethality
Describes a situation in which mutations in two genes together result in cell death, but a mutation in either gene alone does not.
Tetrad analysis
A tetrad is a group of four haploid spores (ascospores) produced by one meiotic event, followed by mitosis. In organisms like yeast, the four spores are encased in a single sac called an ascus.
Purpose of tetrad analysis
Determine whether two genes are linked (i.e. on the same chromosome).
Map the distance between genes.
Distinguish between recombination and independent assortment.
Study gene conversion and meiotic segregation patterns.
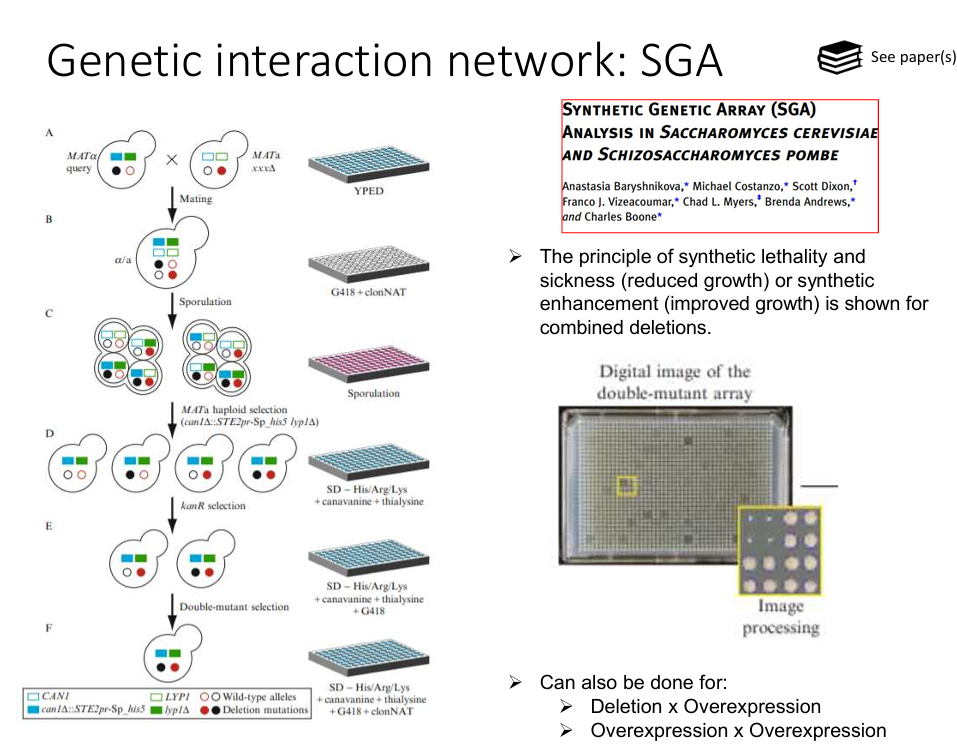
Synthetic Genetic Array (SGA)
Synthetic Genetic Array (SGA) analysis is a high-throughput technique developed in Saccharomyces cerevisiae (budding yeast) to systematically investigate genetic interactions.
It is done by generating double mutants between a query mutation (gene of interest) and a large collection of deletion mutants (typically the whole genome), and observing their phenotypes.
How does it work?
Start with two strains:
A query strain carrying a specific mutation (e.g. gene A deleted)
A library of deletion mutants (each lacking a different non-essential gene)
Mating:
The query strain is mated with each strain in the deletion collection to form diploids.
Diploid Selection:
Use selective media to select for only diploids that contain both mutations.
Sporulation:
Induce meiosis and sporulation in diploids to generate haploid spores.
Haploid Selection:
Select for haploid progeny that carry both the query mutation and the deletion from the library.
Growth Analysis:
Analyse growth of double mutants.
Poor or no growth = negative genetic interaction (e.g. synthetic lethality, the two genes are in redundant/parallel pathways).
Enhanced growth = positive genetic interaction (i.e. one mutation supresses the other’s defect).
Enrichment screening
Experimental approach used to identify mutants that have a growth advantage or survival benefit under a specific condition (e.g. drug exposure, nutrient limitation, or stress).
How It Works
Start with a mutant deletion library: Each mutant has a different gene deletion.
Apply selective pressure:
Grow the entire pool under a condition that challenges cell fitness, e.g. drug treatment, temperature stress, nutrient limitation.
Serial passaging:
Allow cells to grow and divide for several generations under the condition.
Mutants that grow better will become more abundant ("enriched").
Sample collection:
Take cell samples at the beginning (T0) and after multiple time points (T1, T2, …).
Quantify mutant abundance:
Use barcode sequencing, microarrays, or PCR to measure the relative abundance of each mutant over time.
Data analysis:
Compare barcode counts at different time points.
Identify mutants that increase in abundance → these have a growth advantage.
Chemogenetic screening
Chemogenetic screening uses a library of yeast mutants to identify:
Drug targets (direct binding partners)
Genes that buffer or sensitize the cell to the drug
The cellular pathway affected by the drug
It helps determine a drug’s mechanism of action (MoA) by observing how different gene knockouts respond to the drug.
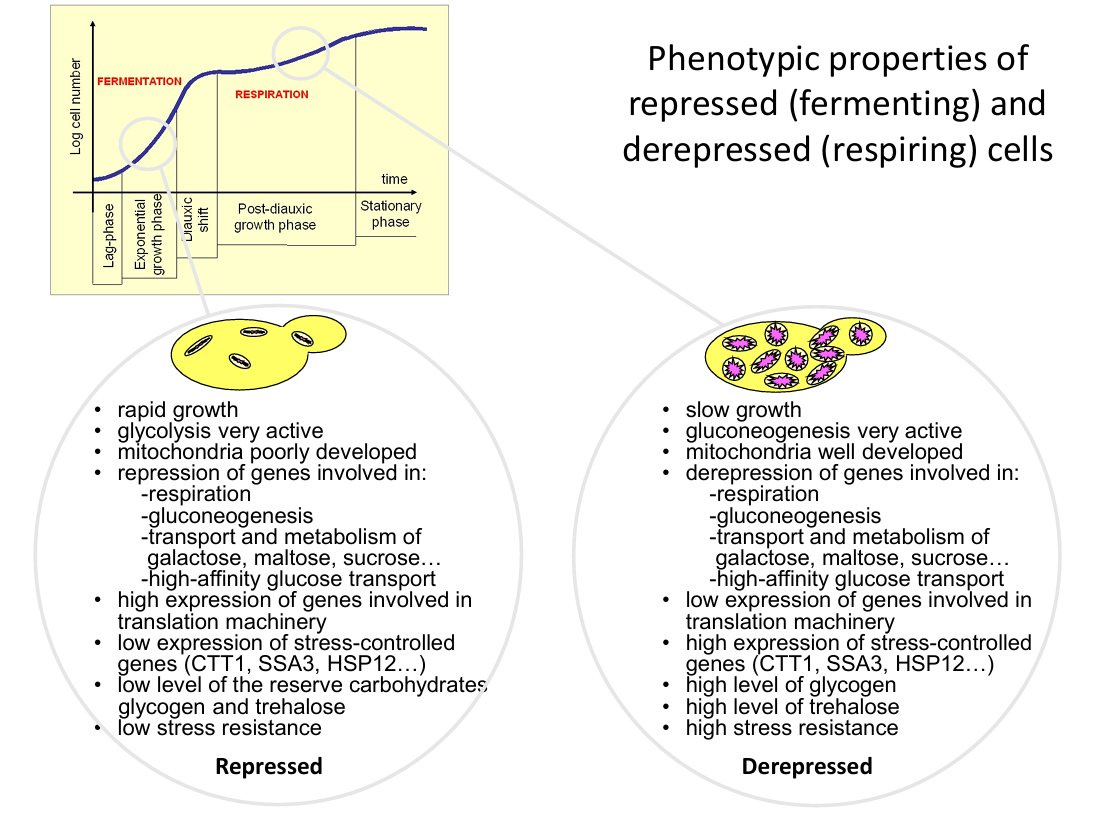
Explain how respiration works in yeast.
Yeast can generate energy through two main processes: aerobic respiration and anaerobic fermentation.
Aerobic respiration (with oxygen, needs mitochondria):
Carbon source, usually glucose, is broken down, and ATP is produced in mitochondria. If glucose is depleted, the other carbon sources can be used, such as ethanol (product of fermentation).
If no oxygen is present, mitochondria cannot be used for respiration and the yeast switches to anaerobic fermentation.
Explain how fermentation works in yeast.
Yeast can generate energy through two main processes: aerobic respiration and anaerobic fermentation.
Anaerobic fermentation (no oxygen, no mitochondria needed)
Glucose is broken down into pyruvate (glycolysis), producing 2 ATP. The pyruvate is then converted into ethanol and carbon dioxide. This is much less efficient than respiration.
What are the advantages of yeast being able to do both respiration and fermentation?
Flexibility in experimental conditions: it allows researchers to manipulate oxygen levels, carbon sources (like glucose vs. ethanol), or stress conditions and study how metabolism, gene expression, and cell signalling adapt (see figure).
Model for mitochondrial biology: Researchers can study mitochondrial function, mutations, and diseases like mitochondrial myopathies, since yeast mutants can survive without respiration (by using fermentation). Thus, even lethal mitochondrial mutations can be studied.
Yeast VS Human mitochondrial genome.
Order of genes is conserved.

Mitochondrial functions are encoded by…
both the mitochondrial and nuclear genomes.
Yeast petite
Yeast petite colonies (also called rho) are much smaller than those formed by wild type cells due to deficiencies that prevent aerobic respiration. On a medium that only support aerobic respiration, petite cells are unable to grow.
What can cause the petite phenotype?
The petite phenotype can be caused by:
the absence of mitochondrial DNA (rho0)
mutations in mitochondrial DNA (rho-)
mutations in nuclear-encoded genes involved in oxidative phosphorylation (aerobic respiration).
rho+
Yeast strains with wild type mitochondrial DNA or mutants having altered mitochondrial DNA that still results in functional mitochondria.
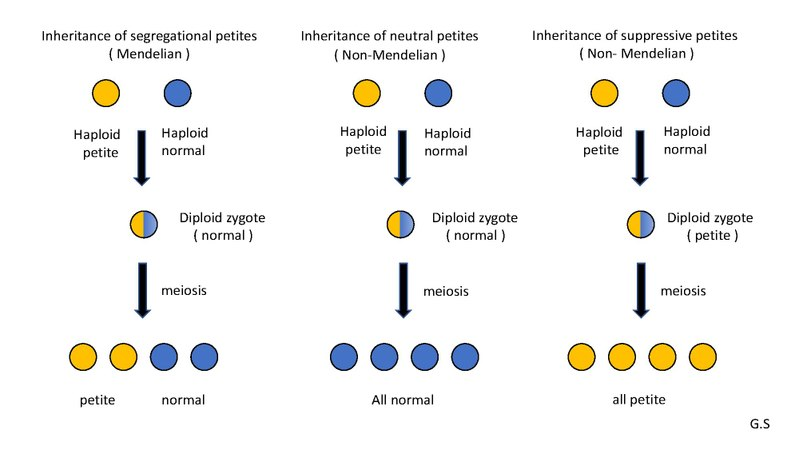
Three main types of petite mutation inheritance in yeast.
Reminder: “Petite” mutants arise from defects in mitochondrial DNA (mtDNA) or nuclear genes required for mitochondrial function. They cannot perform aerobic respiration, so they grow more slowly by fermentation, resulting in small colonies.
Types of inheritance:
Segregational petites (pet-):
Cause: mutation in a nuclear gene.
Inheritance: Mendelian 1:1 segregation in meiosis.
Neutral petites (rho-N):
Cause: mutation in mtDNA, but non-interfering.
Inheritance: when crossed with wild type, all offspring are wild-type.
Supressive petites (rho-S):
Cause: mutation in mtDNA, and interferes with normal mtDNA replication.
Inheritance: when crossed with wild-type, all offspring are petite.
Most petit mutants of S. cerevisiae are of the … type.
supressive
Strains
Two different strains are the same species, can produce viable offspring, but have different properties.
For example, there are hundreds of strains for wine making, leading to different end products.
Biotechnology applications of yeast
Synthetic biology: building pathways to create an end product (e.g. carburant, medicine, beer, food…) using yeast as a host cell. Toolkits such as the MoClo-Yeast toolkit exist to facilitate the design of such processes.
CRISPR-Cas: can be used to modify promoters to increase or lower expression.
Forced laboratory adapted evolution: controlled experimental process where microorganisms (like Saccharomyces cerevisiae) are grown under specific, often stressful conditions for many generations, forcing them to evolve mutations that help them survive or grow better in that environment.