T or F: Regulatory strategy aligns the regulatory and compliance activities with a business strategy to bring a new or modified product tomorrow in the desired regions within the desired timeframes
True
T or F: an important part of regulatory strategy is determining the regulatory pathway in key milestones and timelines associated with that pathway
True
T or F: As part of regulatory intelligence, a Regulatory Affairs professional, does not need to concern himself slash herself with the business objective or problem the medical device aims to solve
False
T or F: It is important to be aware of the regulatory Pathways and timelines that your business competitor products have used
True
T or F: the medical devices field is a regulated field, meaning certain rules must be followed in order to manufacture and Market medical devices
True
T or F: it's important to get buy-in from critical stakeholders for your projects regulatory strategy
True
T or F: you are working on a medical device in your company and realize that it treats a condition/disease that is rare. You should consider a request for a humanitarian device exemption from FDA as part of your regulatory strategy
True
T or F: determining whether an IDE in clinical studies are required is an important part of us regulatory strategy
True
T or F: GDP, GLP, and GMP are all part of quality compliance requirements
False
T or F: an important part of regulatory strategy is determining whether to submit a pre-submission meeting request with FDA (Q-sub)
True
T or F: a solid regulatory strategy is one of the foundations upon which successful medical product development is based
True
T or F: a citizen petition is where FDA holds a workshop requesting industry to take or refrain from taking certain actions
False
T or F: your company has submitted a q sub requesting a meeting with FDA. FDA must provide feedback within 90 days of receipt or 3 days before scheduled meeting, whichever is sooner
False
T or F: it's a good idea to ask FDA open-ended questions so that FDA can tell you what they would like you to do
False
T or F: Nancy Morrison shared in class about a class 3 device that measured glomerular filtration rate for determining kidney health. The device was classified as a breakthrough therapy device
True
T or F: the above mentioned device was submitted at the traditional PMA, where all the data/testing is / was submitted at once
False
T or F: all medical device manufacturers must follow FDA guidance, which has the force of law
False
T or F: the intended use encompasses the more specific indication for use of a device
True
T or F: your company has submitted a 510k. FDA has identified some deficiencies and put the submission on hold and has requested additional information/data. However, it's unclear on exactly what the problems are or what FDA would like to see. You submit a submission Issue request requesting to me to discuss the deficiencies. FDA must grant the meeting within the next 3 weeks
True
T or F: a study test determination request is a request submitted to your IRB to determine whether you need an IDE for your device
False (FDA)
T or F: for your company's PMA, you can request a PMA day 120 meeting to discuss the status with FDA at submission day 120
False
T or F: devices not approved or cleared by FDA may be exported from the US
True
Which of the following would not be included in a regulatory plan?
1) scope
2) regulatory pathway selected and why
3) possible or anticipated inspections
4) intended use or indication for use
5) none of the above
None of the above
Your company's planning on submitting multiple submissions to FDA over the next 6 to 12 months. You are concerned that the FDA should understand some of the nuances in your devices performance or technology. You recommend that your company do the following:
1) request an information meeting with FDA
2) request a study risk determination
3) request an FDA Workshop
4) submit a submission Issue request
5) request an evidentiary hearing
Request an information meeting with FDA
Which of the following is not sound advice for a meeting with FDA:
1) learn about the FDA Personnel who will attend the meeting
2) attempt to anticipate FDA's position
3) write a proposed agenda for the meeting
4) seek specific answers and commitment from FDA to achieve your meeting goal
5) be defensive and wary, and don't communicate too openly
6) none of the above
Be defensive and wary, and don't communicate too openly
Which of the following would not be a part of GMP?
1) a procedure for maintaining Version Control of documentation
2) a procedure for reviewing the impact of changes to design, documentation, or processes
3) a procedure establishing design reviews
4) a work instruction describing how a component of the medical device is manufactured
5) none of the above
None of the above
Which of the following is another name for a lot or batch record?
1) device Master record (DMR)
2) device history record (DHR)
3) design history file (DHF)
4) IQ/OQ/PQ
Device history record (DHR)
Which of the following is not an example of a design input?
1) design/assembly drawing
2) performance characteristics
3) reliability requirements
4) sterility requirements
5) handling and storage requirements
Design/assembly drawing
In the eyes of FDA, who is ultimately responsible for ensuring GMP regulations are established and implemented?
1) design engineers
2) quality engineer
3) executive management
4) regulatory professionals
Executive management
Which of the following is not a part of a recall strategy?
1) level of Effectiveness check
2) public warning needed
3) CAPA
4) recall death
CAPA
Which of the following is not a level of Effectiveness check for a recall?
1) level A - 100% of consignees
2) Level B - 75% of consignees
3) Level B - 50% of consignees
4) Level C - 10% of consignees
5) Level D - 5% of consignees
6) Level E - no Effectiveness check required
Level D - 5% of consignees
Warning letters are public and are typically escalated
1) establishment inspection reports
2) 483 observations
3) 522 observations
4) red howler letters
483 observations
Which of the following is not an example of quality by Design
1) testing the product
2) establishing user needs and ensuring the medical device meets those needs
3) design changes are reviewed and approved in their impacts determined in design reviews
4) specifications, manufacturing procedures, and work instructions are reviewed, approved, and released to manufacturing as part of design transfer
Testing the product
A pre-submission to the FDA should contain:
1) device description
2) the proposed intended use / indication for use
3) specific questions
4) propose date/times for meeting
5) all of the above
6) none of the above
All of the above
Circle the following statements that are not true regarding the 510k program:
1) 510k submitters must pay a user fee to FDA
2) a 510k is an application to FDA that claims that your device is substantially equivalent to the predicate device and therefore at least a safe and effective as that device
3) multiple predicates always make it easier for FDA to determine substantial equivalence
4) the FDA review clock for a traditional 510k is 90 days
multiple predicates always make it easier for FDA to determine substantial equivalence
Which of the following is an example of a biocompatibility test that FDA will require for device that have long-term impact with blood, tissue/bone / Dentin but it's not likely to be required for shorter-term contact devices:
1) carcinogenicity
2) cytotoxicity
3) irritation or intracutaneous reactivity
4) sensitization
Carcinogenicity
The results of a design effort at each design phase (ex. Dimensional specifications, sterility, Packaging)
Design output
Process by which the equipment's ability to perform the function for which it was designed is verified
Operational qualification (OQ)
The process by which a design specifications are transferred to production
Design transfer
Process by which the performance of the equipment and the actual manufacturing process is validated
Performance qualification (PQ)
The compilation or index that refers to all of the instructions, drawings, procedures, and records that are used to manufacture the device. The device recipe
Device Master record (DMR)
A document that describes the regulatory strategy for a particular product
Regulatory plan
The process of determining the objective evidence whether the design outputs match the design inputs. This ensures that the product is made correctly. Internal testing
Verification
Initiates a process to address a non-conformity that has occurred to prevent the failure from happening again
Corrective action
A collection of the documents that describe the design and development of a medical device. It's purpose is to demonstrate that design controls were followed in the design, development, and manufacture of the device
Design history file (DHF)
The lot or batch record of a specific lot or batch of a devices production history. It indicates the manufacture date, the progress of the device through the manufacture steps, contains copies of the devices labeling, and indicates the number of devices passing inspection that were released to Market in that lot / batch
Device history record (DHR)
Qualification and calibration of equipment when installed
Installation qualification (IQ)
The process of determining by objective evidence whether the device meets its intended use and function. This ensures that the right product was made and that it meets user needs. External testing. This is performed on the entire device or system (simulated use conditions) and not just individual components
Validation
A company's removal or correction of a marketed product that FDA considers to be in violation of the laws and administers and against which the agency would initiate legal action, seizure
Recall
Initiates a process to address a trend or potential issue in order to prevent the failure from occurring
Preventive action
Any product not meeting specifications
Non-conforming product
Repair, modification, adjustment, re-labeling, destruction, or inspection of a product without its physical removal to some other location
Correction
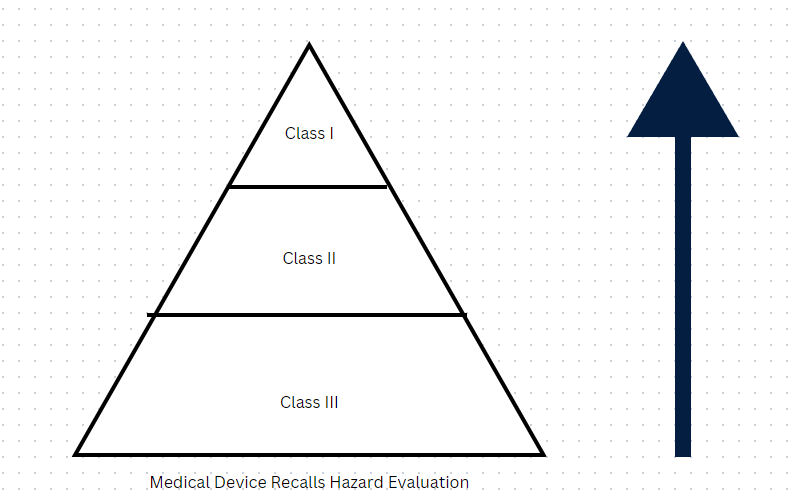
Use of or exposure to the device will cause serious adverse Health consequences or death (what type of recall)
Class 1 recall
Administrative review for a refuse to accept: FDA has ____________ days to indicate whether a submission contains all the necessary parts
15
QMS documents are said to be controlled documents what does this mean?
Versions of the documents are recorded
What is a quality management system and what is its purpose?
a QMS is a system established by a company to ensure a device is made correctly and reduced the amount of errors and mistakes in production and after the device has hit the market
This class of recall is not likely to cause adverse Health consequences
Class 3
Manufacturers must submit reports to the FDA no later than _______ calendar days after the date the manufacturer receives or becomes aware of information that suggests a device it markets: may have cause or contributed to a death or Serious injury or has malfunctioned and this device or a similar device in markets would likely cause or contribute to a death or Serious injury if the malfunction were to occur
30
A correction or removal of a distributed device that involves a minor violation of the act that would not be subject to legal action by FDA or that involves no violation of the act is known as a
Market withdrawal
In order to legally export a legally marketed medical device out of the US list one of the following requirements that must be met
1) registered with the FDA
2) legal requirements
3) manufacturing GMP
An example of a device user facility in terms of reporting requirements is a
Hospital
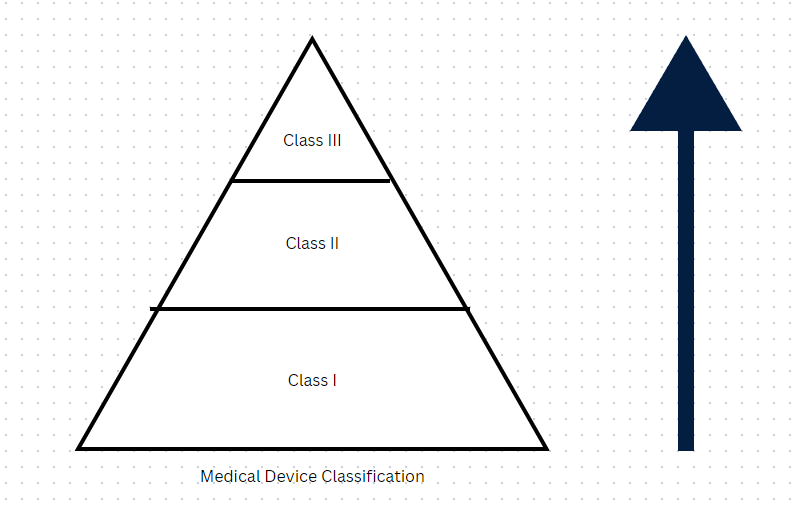
Fill out the following pyramid:
Fill out the following pyramid:
A unique device identifier is composed of two parts. The first part is called the ________________________, and gives information about who is the ___________________ and Associates the device with a number. The second part is called the ______________________________ and gives information like (name one example): ___________________________
Device identifier, manufacturer, production identifier, what batch it's from
For each complaint a manufacturer receives, the manufacturer must determine whether to ___________________________ and if an _________________________ is necessary
Report to the FDA, investigation
Name an important procedure that must be implemented as part of a QMS
Control documents, a procedure for dealing with non-conforming devices
T or F: the work of muckrakers was influential in passing legislation that was the precursor to Modern regulations
True
T or F: in 1906, the head of the FDA, Harvey Wiley organized the poison Squad which sought to expose 12 healthy volunteers to known poisons
False
T or F: the 1906 Pure Food and act included regulations for medical devices
False
T or F: Upton Sinclair's the jungle made the country aware that the country's meet was probably not safe
True
T or F: sulfanilamide is a poison that killed over 100 Americans in 1937
False
T or F: one of the reasons that the sulfanilamide poisoning happen was that no safety testing was performed on the liquid version
True
T or F: the 1906 Pure Food and Drug Act gave FDA the ability companies for a criminal violations of the ACT
True
T or F: outrage due to beaches of assumed trust often leads to new regulations
True
T or F: the medical device user fee in modernization act sought to reduce the FDA review bottleneck by through industry fees for submissions to FDA in exchange for FDA agreements to meet review timelines
True
T or F: The coronavirus Aid, relief and Economic Security (CARES) Act which was passed during the pandemic requires that manufacturers must report shortages of all devices
False
T or F: the 510k program is intended to show that devices are substantially equivalent, or in other words, identical, to a pre-existing device on the market
False
T or F: If there are multiple devices that could serve as your predicate, in general, it's good practice to include those multiple predicates for your 510 case submission
False
T or F: your company is developing a new pacemaker. A competitor recently received FDA approval for their pacemaker. Because your pacemaker is substantially equivalent to your competitors, you could submit a 510k in order to Market your device and save on development costs, Etc
False
T or F: most class one devices and some Class 2 are 510k exempt, meaning that pre-market notification is not required for those devices
True
T or F: a substantial equivalent decision by FDA in regard to your 510k is considered FDA approved
False
T or F: A pre-market approval by the FDA can be a potential Legal Shield in terms of device liability
True
T or F: then intended use encompasses the more specific indication for use of a device
True
T or F: you can request meetings with the FDA in order to gain insight into the regulatory pathway, IDE protocols, clinical testing required, Etc. via the Q sub program.
True
T or F: if the subject and predicate device of your 510k are devices that you manufacture, you could potentially submit as a special 510k and have a faster FDA review clock of 30 days
True
T or F: FDA does not perform 510K pre-clearance facility inspections, but does perform pre-approval inspections for PMAs
True
The food drug and cosmetics act finally gave the FDA some ”teeth” to ensure companies abided by the law, examples of these include:
1) fines, and warnings, and blacklisting
2) federal court injunctions, product seizures, Factory inspections, and criminal prosecutions for violating the act
3) requiring testing and Factory inspections
4) none of the above
federal court injunctions, product seizures, Factory inspections, and criminal prosecutions for violating the act
The medical device amendments were passed in this year:
1) 1976 and were the first time FDA clearly distinguished between advice and a drug
2) 1970 and required adequate and well-controlled clinical studies prior to marketing for devices
3) 1978 and found that in the previous 10 years there were 10,000 injuries due to medical devices and 731 deaths
4) none of these years
1976 and were the first time FDA clearly distinguished between advice and a drug
Medical device manufacturers are responsible to prove the following when submitting regulatory applications:
1) that the device isn't high risk
2) that the company has a quality system in place and the device is safe
3) the medical device is safe and effective and properly labeled
4) only that the medical device is safe
the medical device is safe and effective and properly labeled
What does not describe how FDA defines a medical device?
1) recognize in the National formulary or us pharmacopeia
2) intended for use in the diagnosis of disease or other conditions, or in the Cure, mitigation, treatment or prevention of disease in man or other animals
3) intended to affect the structure or any function of the body of man or other animals
4) doesn't achieve its primary intended purpose through chemical action within or on the body of man or other animals and is not dependent on being metabolized for achieving its primary intended purposes
5) none of the above
None of the above
The 1990 safe medical devices act added the following to the GMP regulations
1) medwatch reporting
2) pre-production design and validation controls
3) user fees
4) pre-market notification
pre-production design and validation controls
Which of the following is not an example of a device's technological characteristics
1) materials
2) design
3) energy sources
4) features: such as software/hardware, density, porosity, Etc.
5) intended use
Intended use
Which is not a possible 510k format/pathway
1) traditional
2) abbreviated
3) priority
4) special
Priority
Prohibited misbranded and adulterated Foods, drinks, and drugs from entering interstate commerce
Pure Food and Drug Act of 1906
Law that indicated manufacturers be required to provide proof new drugs received for their intended use before being placed on the market. Cosmetics and medical devices were regulated for the first time. FDA was given “teeth” with enhanced enforcement Authority
Food, drug, and cosmetic Act of 1938
Were added as part of the safe medical devices act in 1990 to the GMP regulations because a large number of recalls were due to design defects / errors. These are required once a company decides to Begin work to develop a device for a commercialization
Design controls
Food and Drug Administration. Renamed in 1930 from the Bureau of chemistry. Is an agency within the executive branch
FDA
A reviewer for the FDA who refused to authorize the drug thalidomide in the US, despite pressure from the drug company, until further studies were performed showing safety in pregnant women
Frances Kelsey
Chief chemist and Bureau of chemistry, sought to test chemicals and preservatives used in the food industry to discernment if they were safe. He funded the studies himself because the food lobbies were against his efforts. Eventually he was the first head of the Food and Drug Administration
Harvey Wiley
A drug that was approved in many countries but was later found to cause birth defects when taken by pregnant women. The outrage over the drug became influential in having the kefauver Harris amendment passed in Congress in 1962 to strengthen drugs regulation
Thalidomide