L15- Origins of cancer and early detection recap
1/39
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
40 Terms
what are the origins of cancer
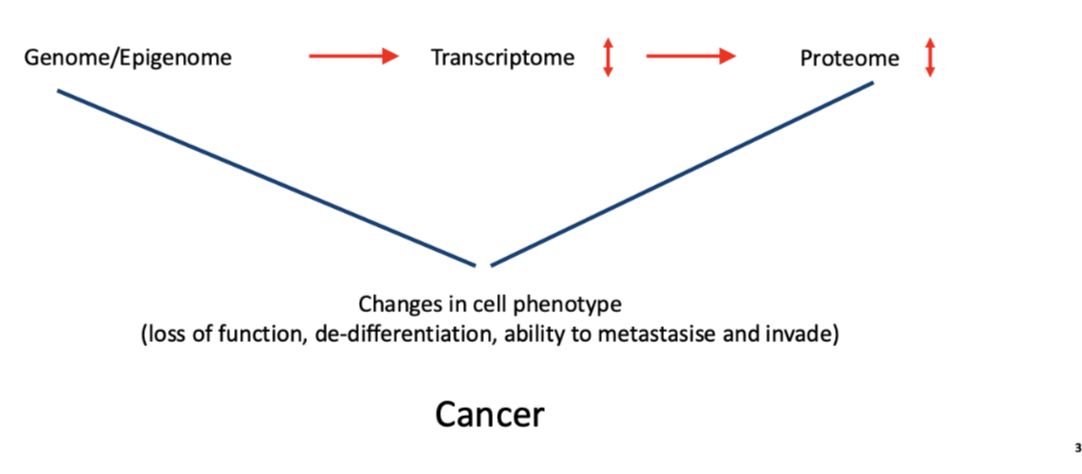
changes in cell phenotype
loss of function, de-differentiation, ability to metastasise and invade
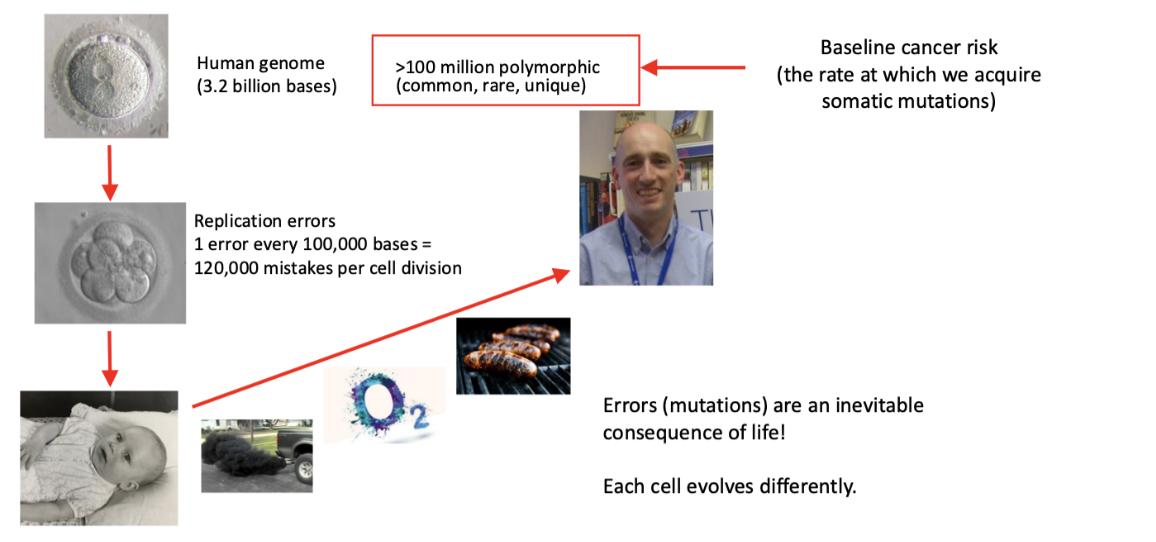
how do constitutional and somatic genomics determine cancer risk
errors/mutations are inevitable
how is cancer a multistep process driven by the acquisition of somatic mutations
spontaneous DNA replication errors
endogenous DNA damage (molecular oxygen)
exogenous DNA damage (UV, ionising radiation, BPDE)
→
DNA damage caused DNA mutation (in TS genes and proto-oncogenes)
→ (genetic instability/antiapoptotic)
cancer
What are high penetrance constitutional genetic variants in cancer?
Rare inherited (monogenic) mutations that greatly increase lifetime cancer risk
eg hereditary breast/ovarian cancer (e.g. BRCA genes)
One defective allele is inherited
Second allele becomes mutated/silenced (somatic “second hit”)
Only a small proportion of cancers are due to these mutations
What is the difference between monogenic and polygenic cancer susceptibility?
Monogenic (single gene):
Rare in the population
High penetrance → strong effect on disease risk
Polygenic (multiple genes):
Common in the population
Low penetrance → each gene has a small effect
Most people have polygenic risk, where many low-risk alleles combine
Cancer risk increases gradually with accumulation of multiple low-penetrance variants
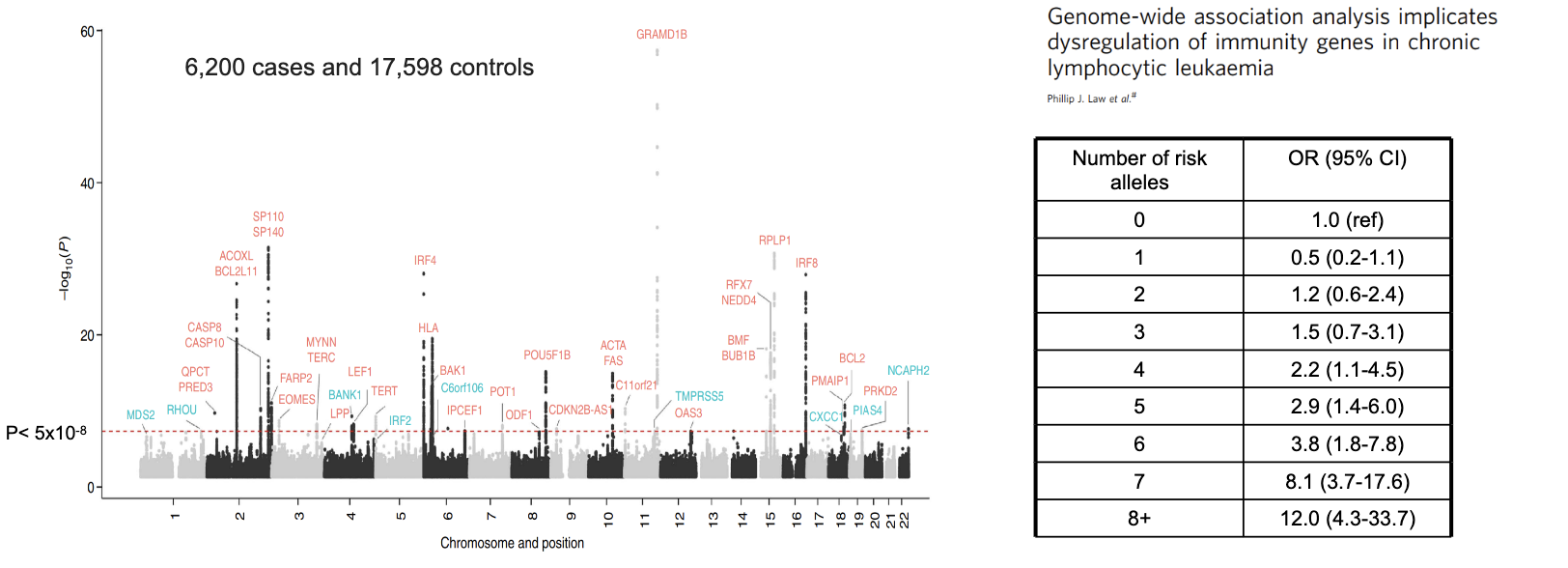
give an example of a polygenic disease
chronic lymphocyte leukaemia (CLL)
>45 loci carrying common variants accounting for 25% of the heritable risk of CLL
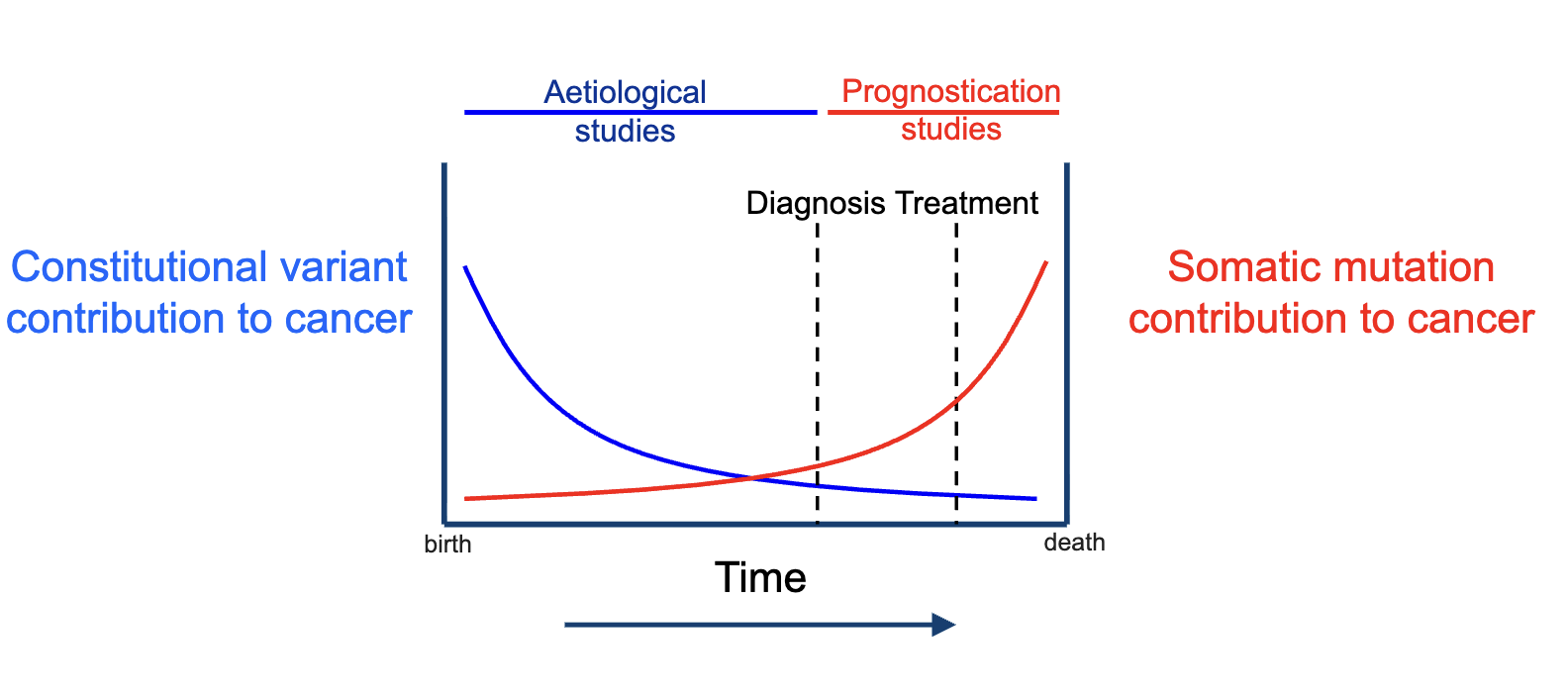
How do constitutional (inherited) and somatic mutations contribute to cancer over time?
Constitutional variants (inherited):
Stronger influence early in life
Declining relative contribution over time
Studied in aetiological studies (causes of cancer)
Somatic mutations (acquired):
Accumulate over time
Increasing contribution as disease progresses
More important around diagnosis and treatment
Studied in prognostication studies (outcomes, progression)
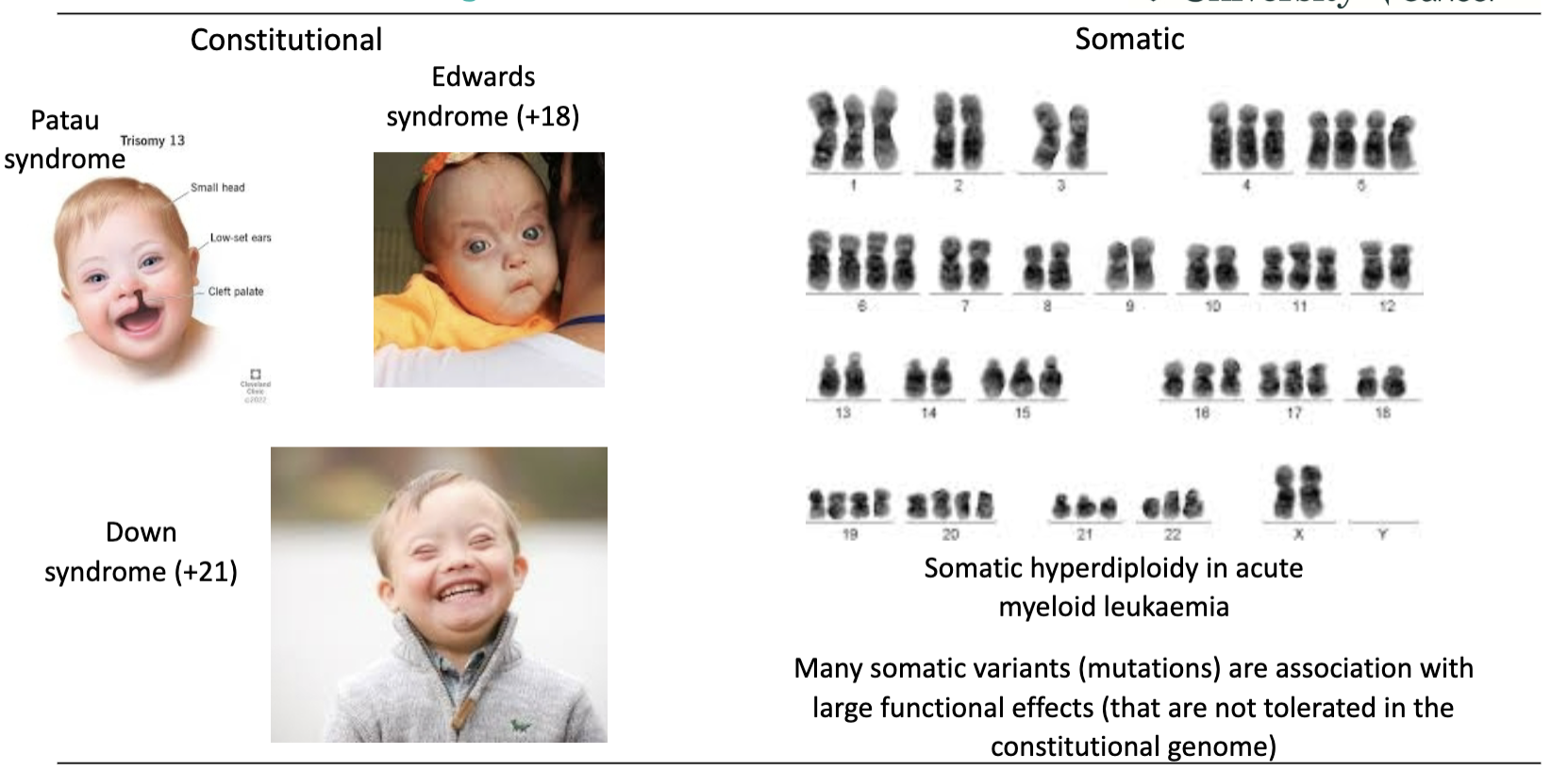
How does tolerance of genetic variation differ between constitutional and somatic genomes?
Constitutional genome:
Less tolerant of genetic variation
Large changes → syndromes (e.g. trisomy 13, 18, 21)
Somatic genome:
More tolerant of genetic variation
Can have large changes (e.g. hyperdiploidy in AML)
Many somatic mutations have large functional effects that are not tolerated in the constitutional genome
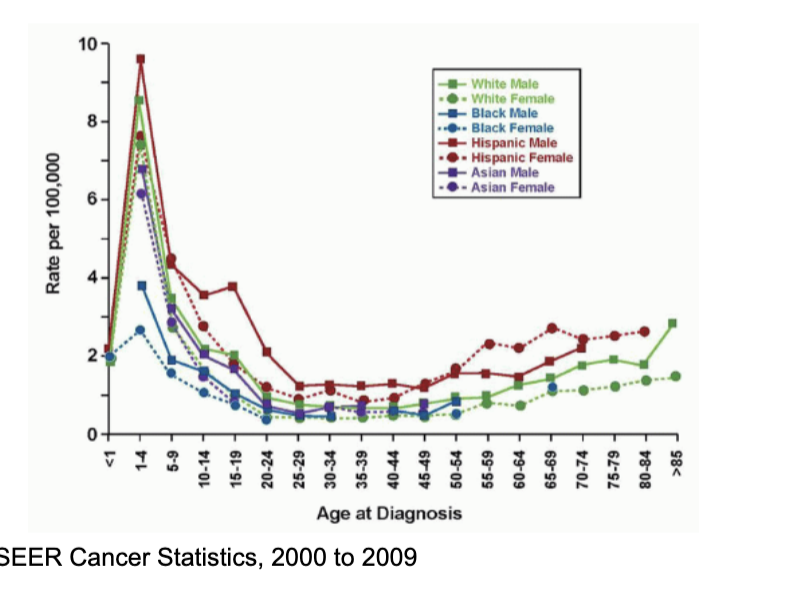
is cancer always an age associated disease
no
eg acute lymphoblastic leukaemia in children
but it is still driven by acquisition and is still affected by exposures
how many somatic mutations are needed for cancer
to 10 driver mutations are needed to cause cancer!
occur in established cancer genes (TP53, MYC)
Major cancer genes are known but many are yet to be identified
Most mutations observed in cancer cells are non-functional passenger mutations (genetic instability)
Identifying driver mutations from passenger mutations is not always easy
give examples of cancers and how many mutations they usually require
1 mutation can cause cancer in some settings (ie. some leukaemias)
Liver cancer requires an average of 4 mutations
Colorectal cancer requires up to 10 mutations
in what ways is cancer a multistep process
genetically
epigenetically
transcriptionally
phenotypically
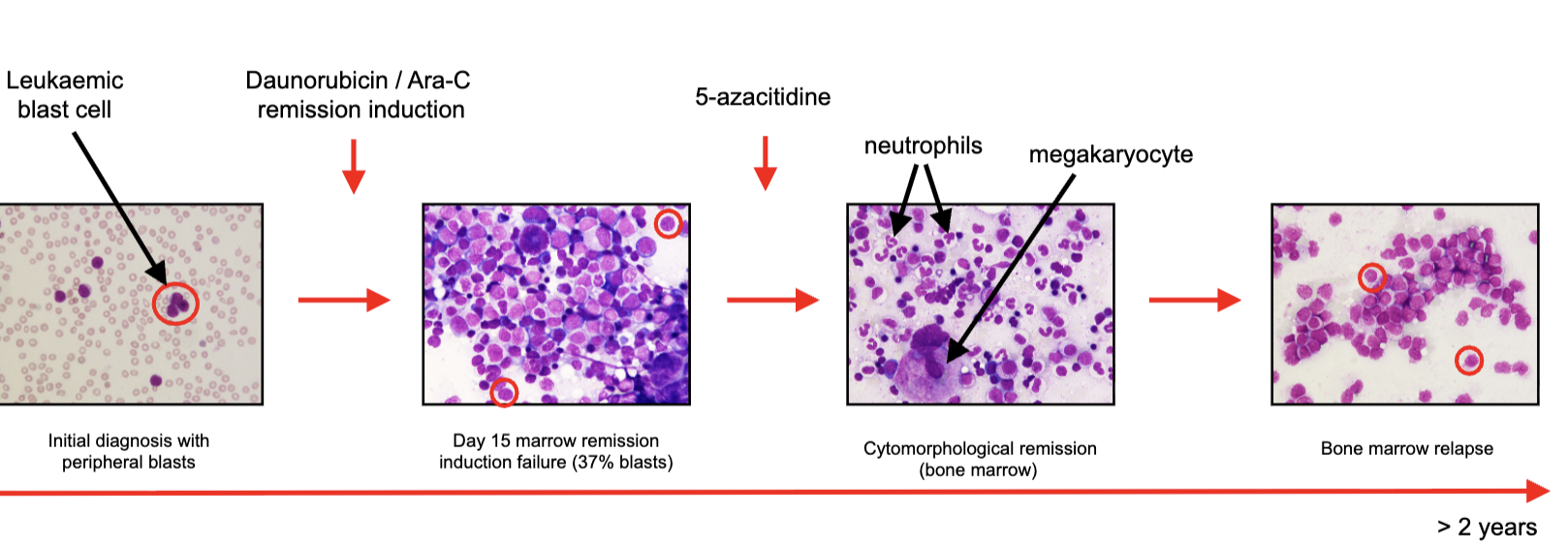
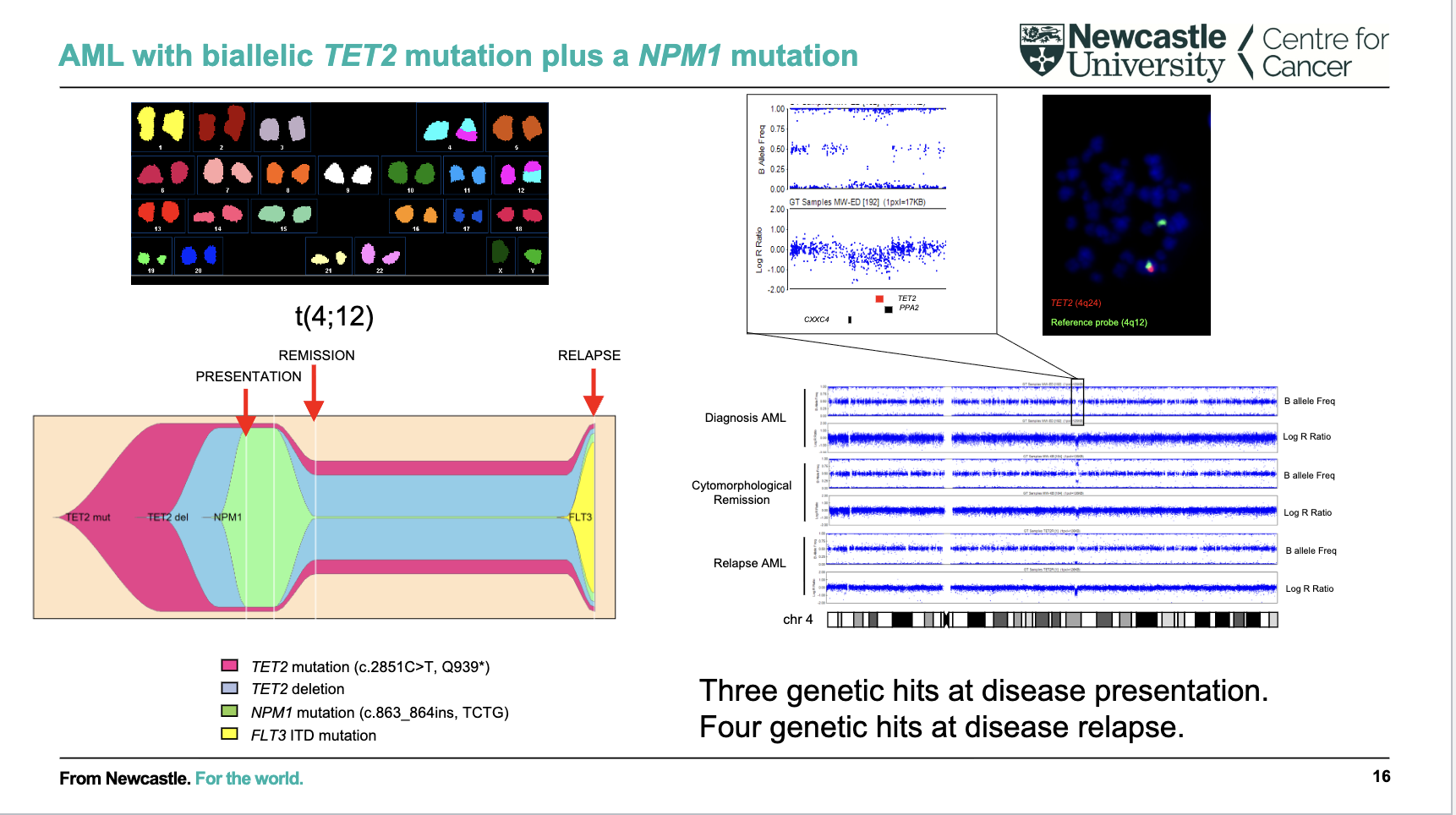
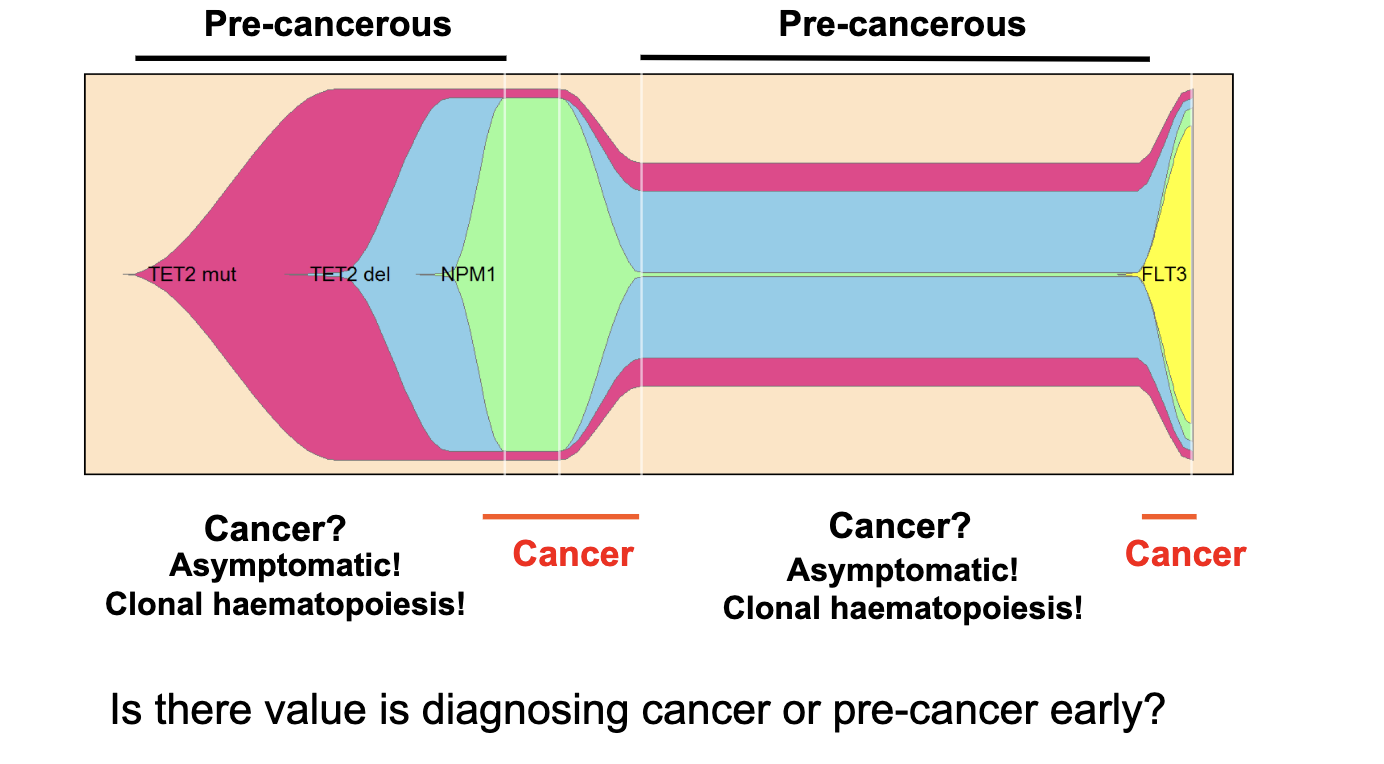
What does this AML example show about mutation accumulation over time?
AML evolves through multiple genetic “hits” (mutations)
At presentation: ~3 key mutations (e.g. TET2 + NPM1)
At relapse: additional mutation acquired (e.g. FLT3)
Shows clonal evolution: cancer changes and becomes more complex over time
Relapse often involves new mutations → disease progression / treatment resistance
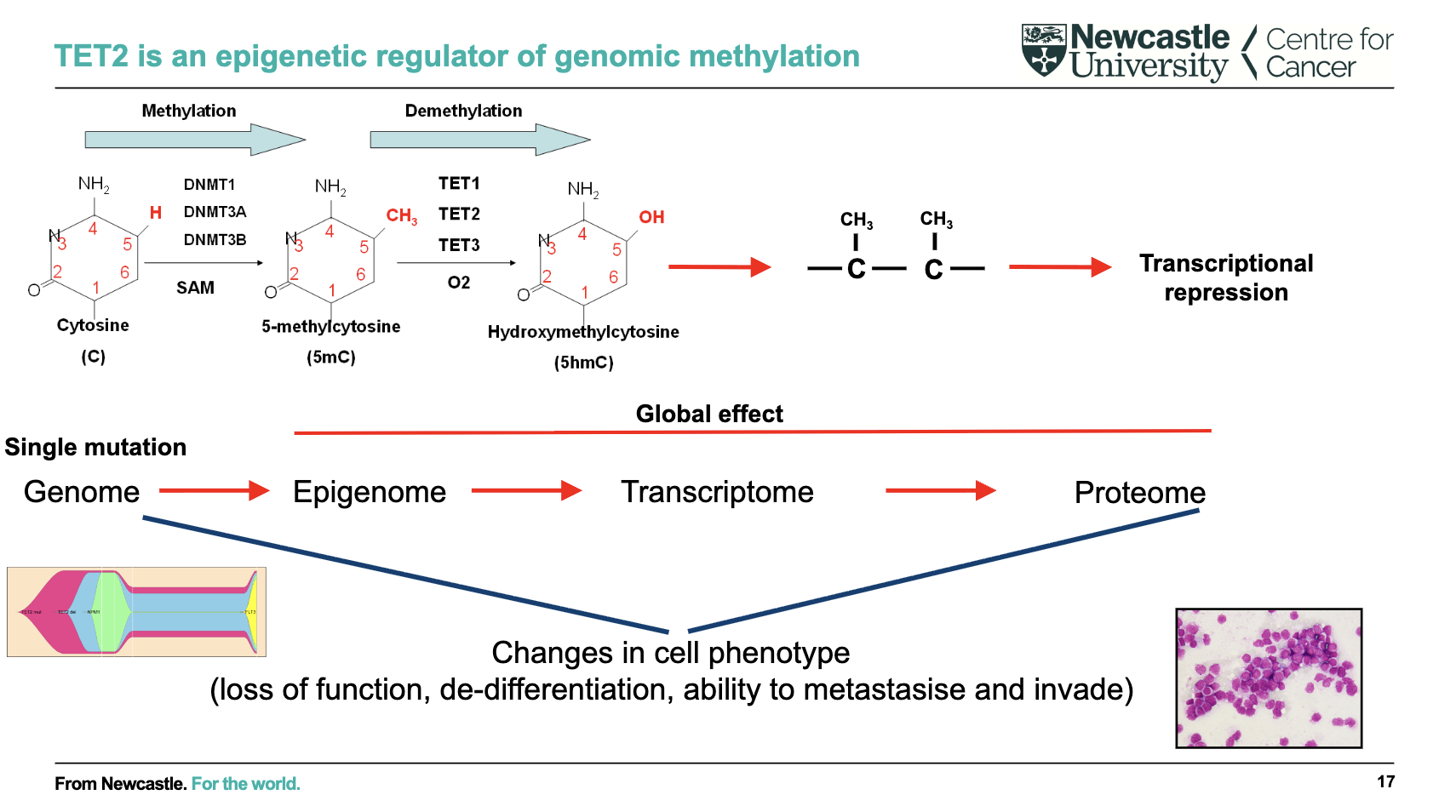
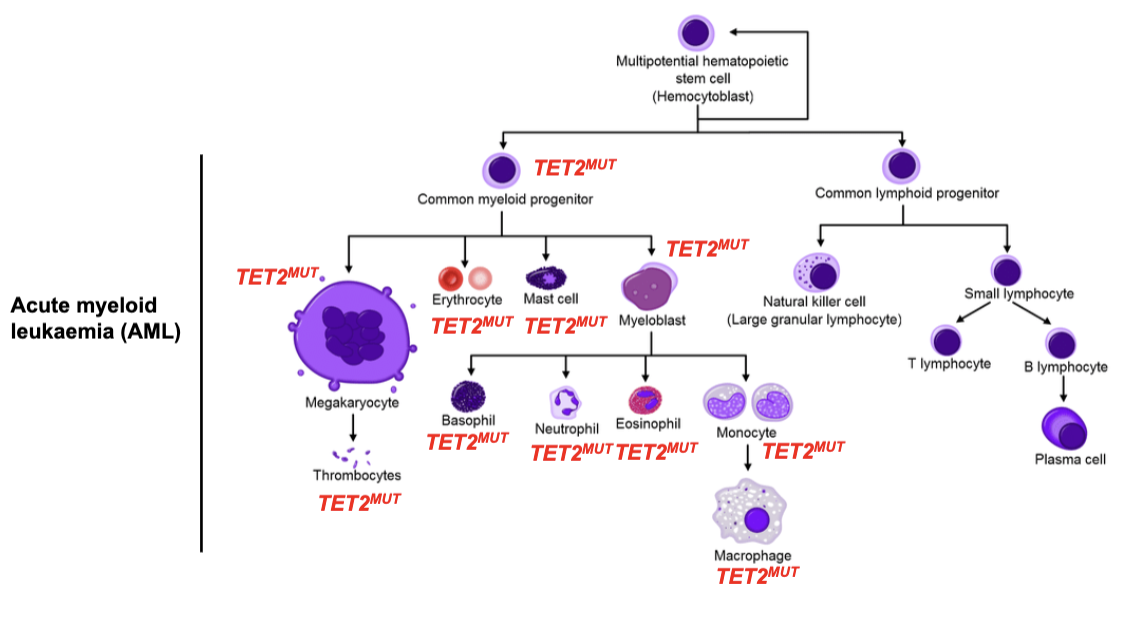
What is the role of TET2 and its global effect on cells?
TET2 regulates DNA methylation (epigenetic control)
Converts methylated cytosine → hydroxymethylcytosine (demethylation pathway)
Methylation → transcriptional repression
Single mutation effect:
Genome → Epigenome → Transcriptome → ProteomeLeads to global changes in gene expression
Results in altered cell phenotype:
Loss of function
De-differentiation
Increased invasion/metastasis
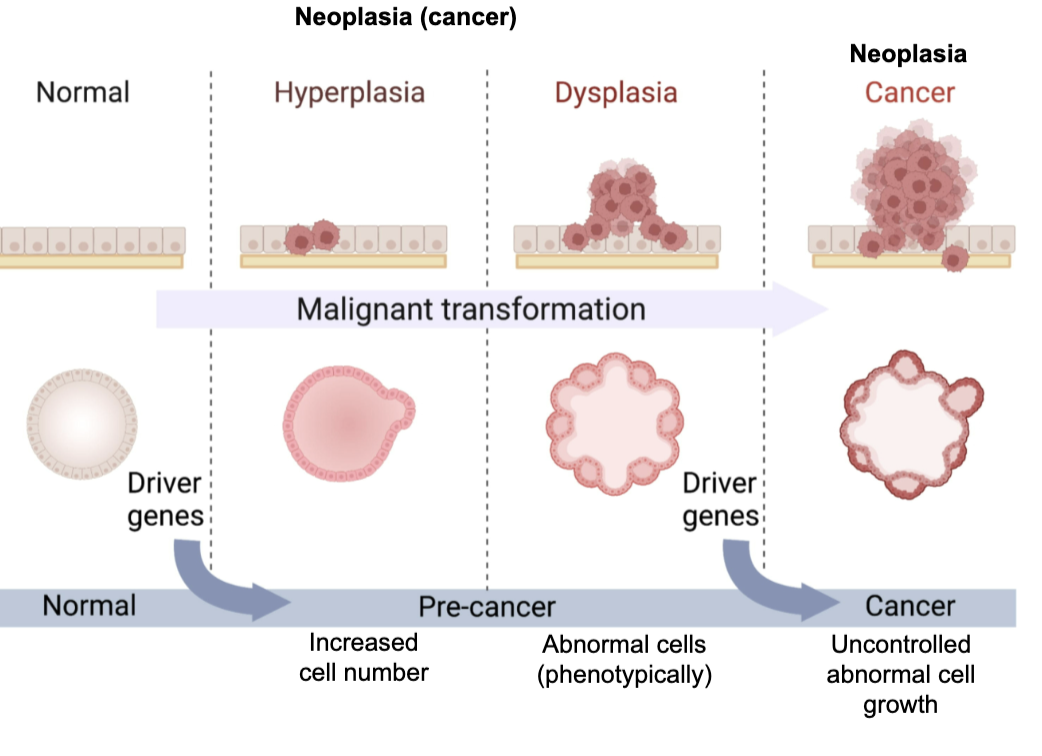
What are the stages of cancer development (malignant transformation)?
Cancer develops as a multi-step process:
Normal → Hyperplasia → Dysplasia → Cancer (neoplasia)Hyperplasia: ↑ cell number
Dysplasia: abnormal cell phenotype (pre-cancer)
Cancer: uncontrolled abnormal cell growth
Driven by accumulation of driver gene mutations
Overall process = malignant transformation
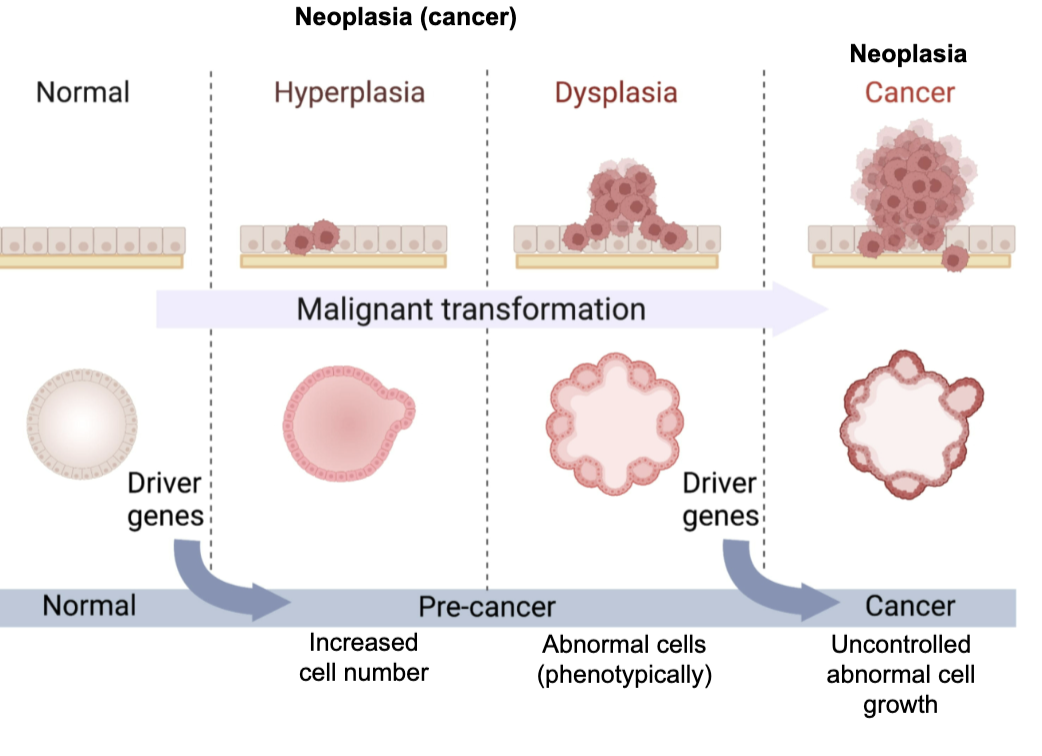
What are key definitions and concepts in neoplasia (cancer)?
Neoplasia = cancer
Benign: cannot invade other tissues
Malignant: can invade other tissues
These terms mainly apply to solid tumours (not myeloid or B-cell cancers)
Haematopoietic cancers:
Derived from non-adherent cells
Disseminate via blood and lymphatic systems
“Acute” vs “chronic” → describes disease aggressiveness
how common is pre cancer
very common
we all have pre cancer as we all have cancer drien somatic mutations
Example: Clonal haematopoiesis - clonal expansion of cells in the blood and bone marrow with leukaemia-initiating mutations.
is clonal haematopoiesis dangerous in all individuals
no- its healthy in asymptomatic individuals
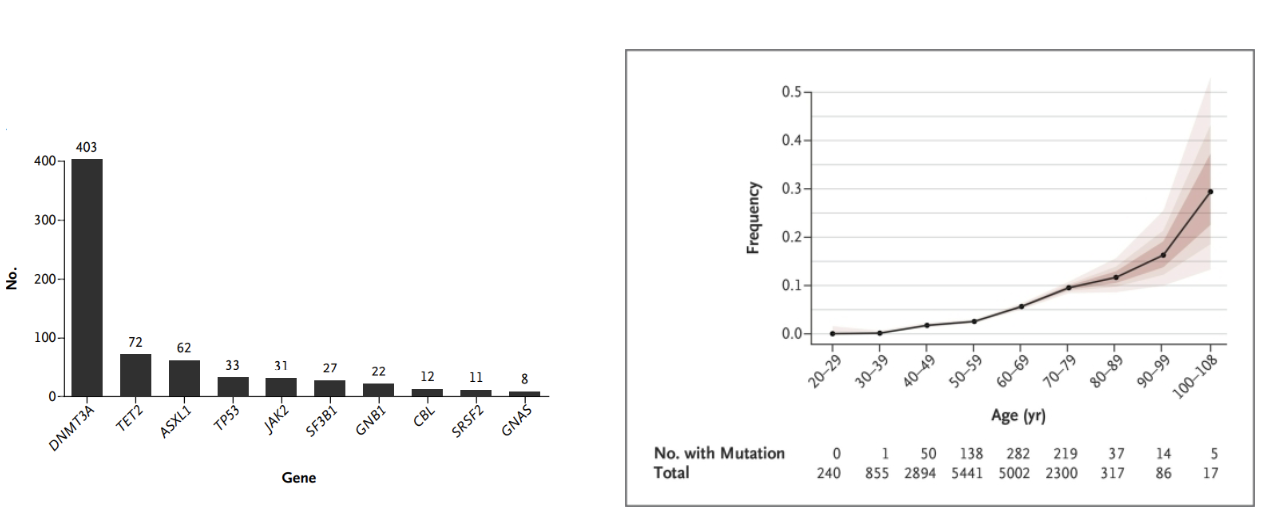
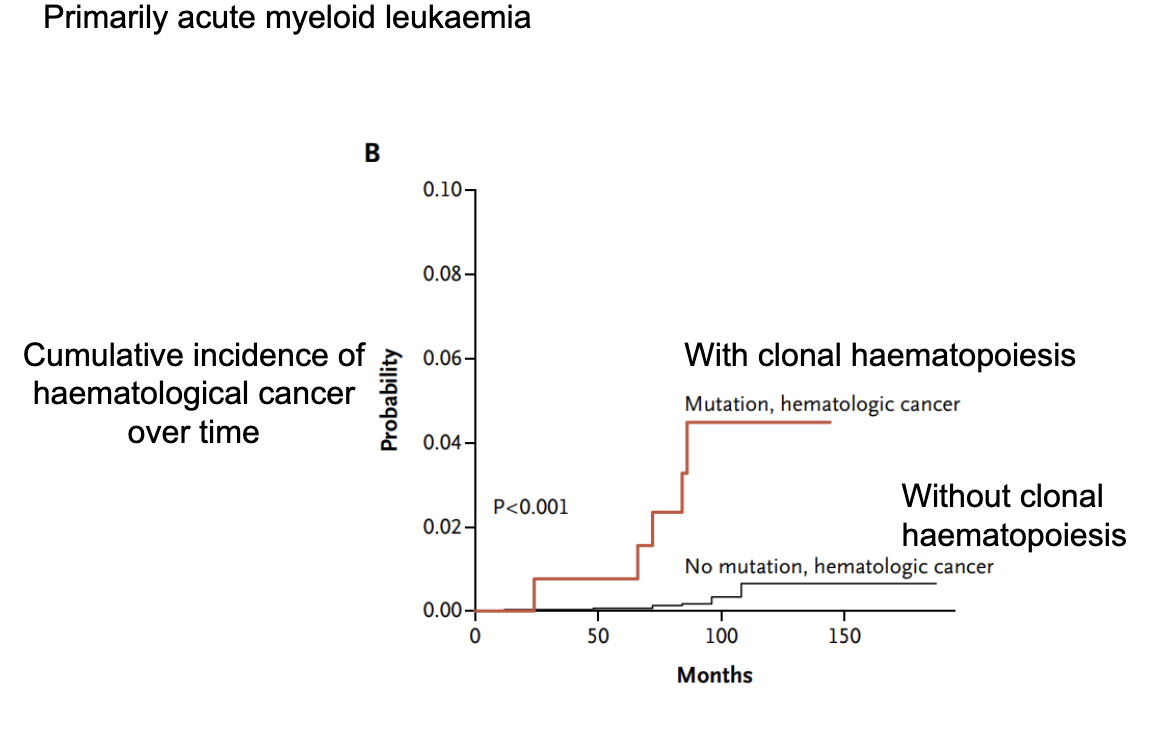
What is clonal haematopoiesis and how does it relate to cancer risk?
Clonal haematopoiesis: presence of blood cell clones with somatic mutations
Associated with increased risk of haematological cancers (especially AML)
Individuals with clonal haematopoiesis have:
Higher cumulative incidence of cancer over time
Significantly greater risk (p < 0.001)
Individuals without mutations have much lower risk
Key idea: early mutations in blood cells can predispose to later cancer development
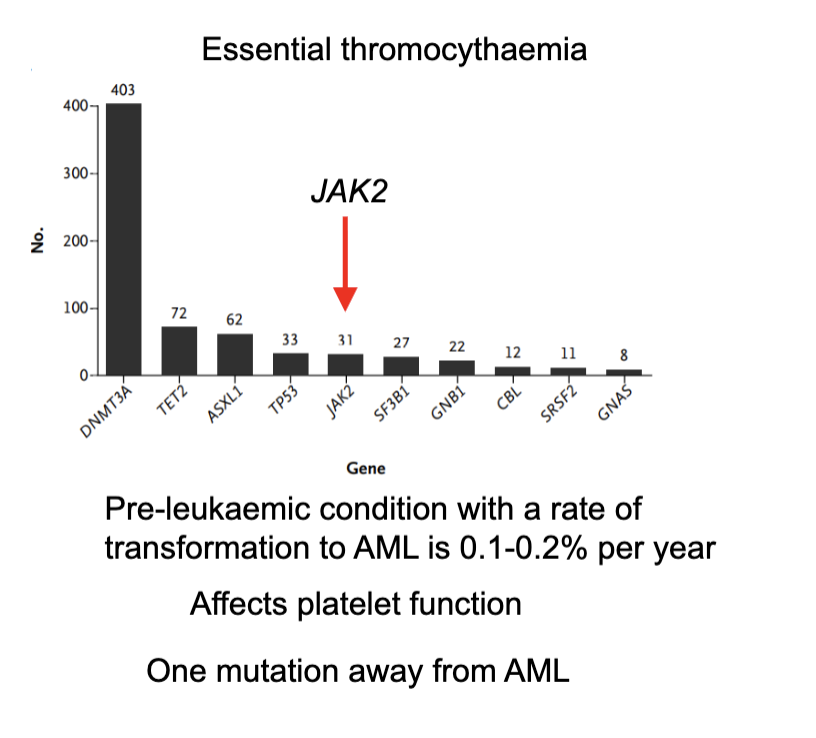
What is essential thrombocythaemia and what are its key features?
A pre-leukaemic blood disorder where the body makes too many platelets
Commonly involves mutations (especially JAK2, among others like DNMT3A, TET2, ASXL1)
Causes abnormal platelet function → risk of clotting or bleeding
Has a low risk of progression to acute myeloid leukaemia (AML) (~0.1–0.2% per year)
Considered close to AML in disease spectrum (“one mutation away”)
when is a cell a cancer cell
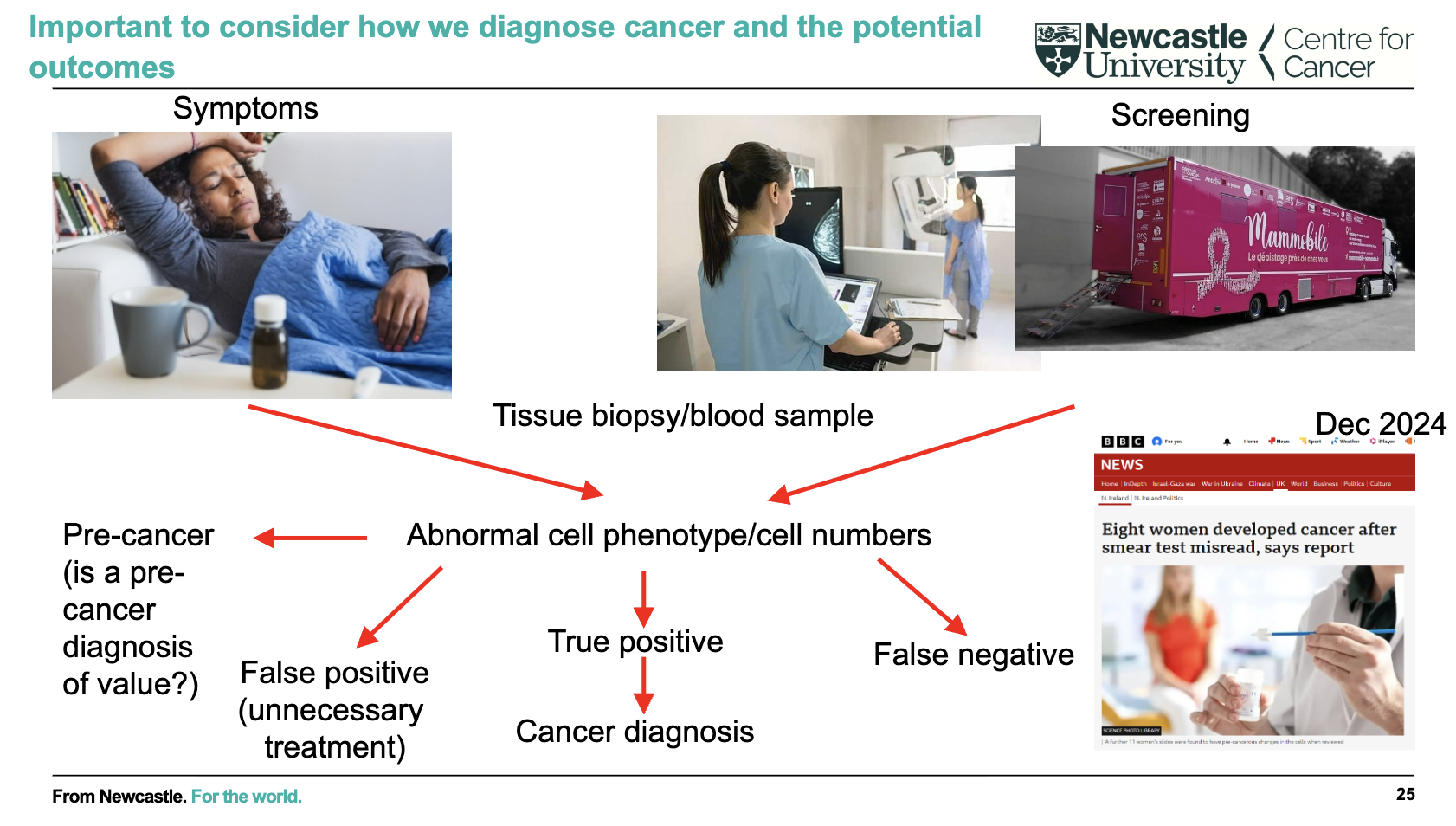
how is cancer screened
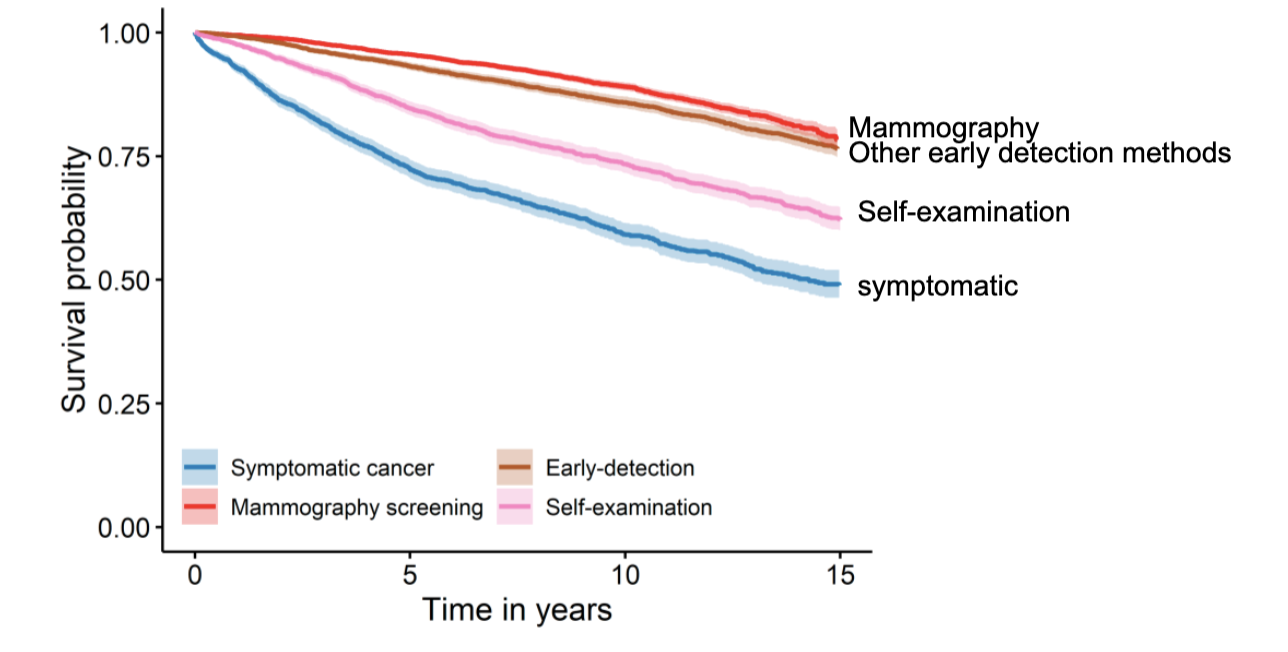
is there a clinical benefit to an early cancer diagnosis
yes for most cases- certainly for many solid cancers
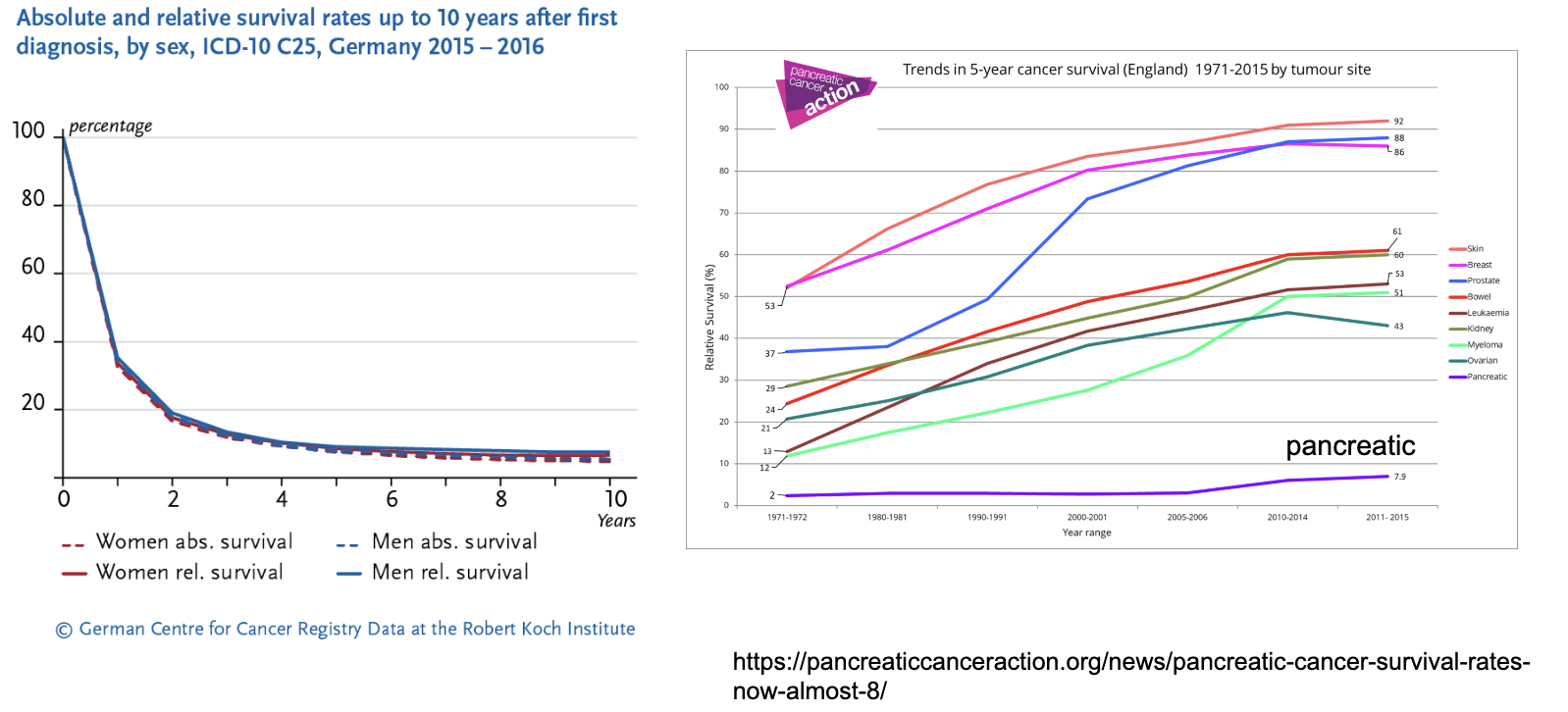
in which cancer is early detection urgently needed
Pancreatic cancer
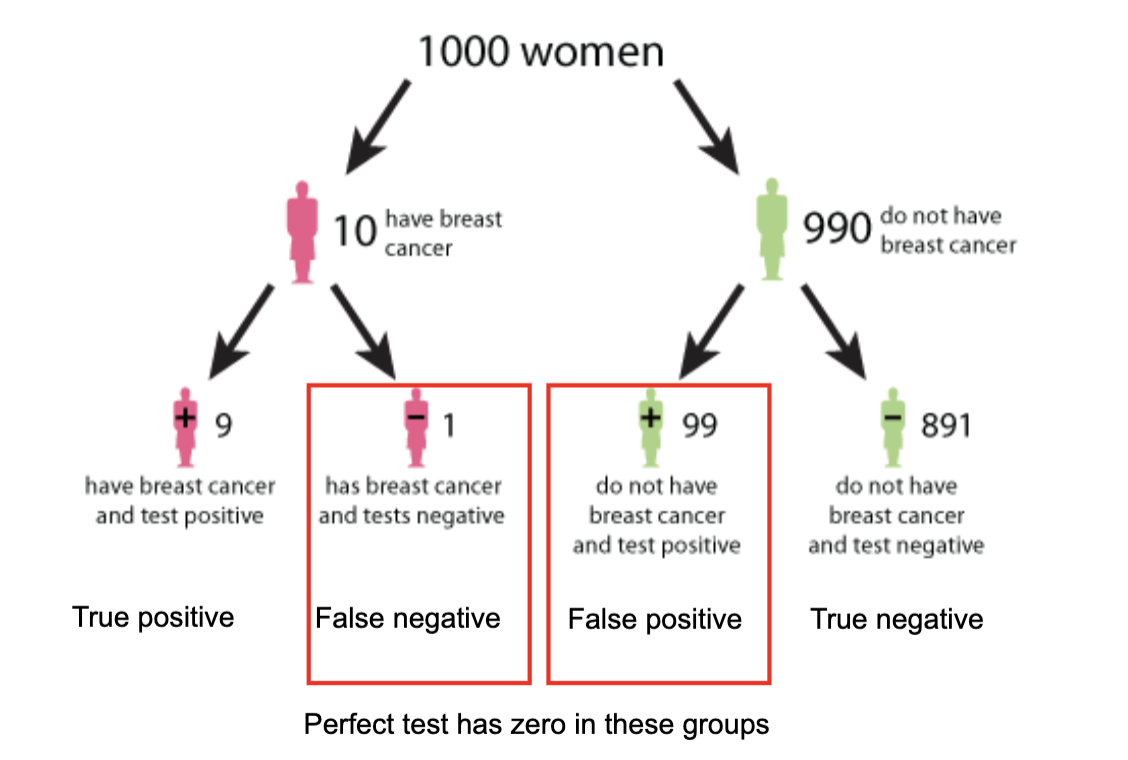
In cancer screening, what are false positives and false negatives, and why do they matter?
False positive: Test says cancer is present when it isn’t → causes unnecessary anxiety, further tests, and cost
False negative: Test misses cancer when it is present → delays diagnosis and treatment
Even with good tests, both can occur (e.g., 99 false positives vs 1 false negative in this example)
A perfect test would have zero false positives and false negatives
Screening decisions must balance accuracy, cost, and practicality
what needs to be considered for tests for cancer diagnosis
Tests for ALL cancers?
Tests for specific cancers?
Cost
Logistics
(easy to perform? painful?)
how can dogs be used in cancer testing
Detects volatile organic compounds (VOCs) generated by cancer cells and exhaled excreted via the lungs in the breath.
Primarily aimed at cancers of the gut (oesophagus, stomach, colon pancreas – plus liver)
what is the prostate specific test
Prostate specific antigen (PSA)
Blood test, painless, cheap!
What does PSA measure and what limits its usefulness as a prostate cancer test?
PSA reflects prostate tissue activity, not cancer specifically
Levels increase with age, even in healthy men
Raised PSA can be due to benign prostatic hyperplasia (BPH), infection, or inflammation
Therefore, PSA is not specific for cancer → risk of false positives
Definitive diagnosis requires biopsy, which is invasive, costly, and carries risks
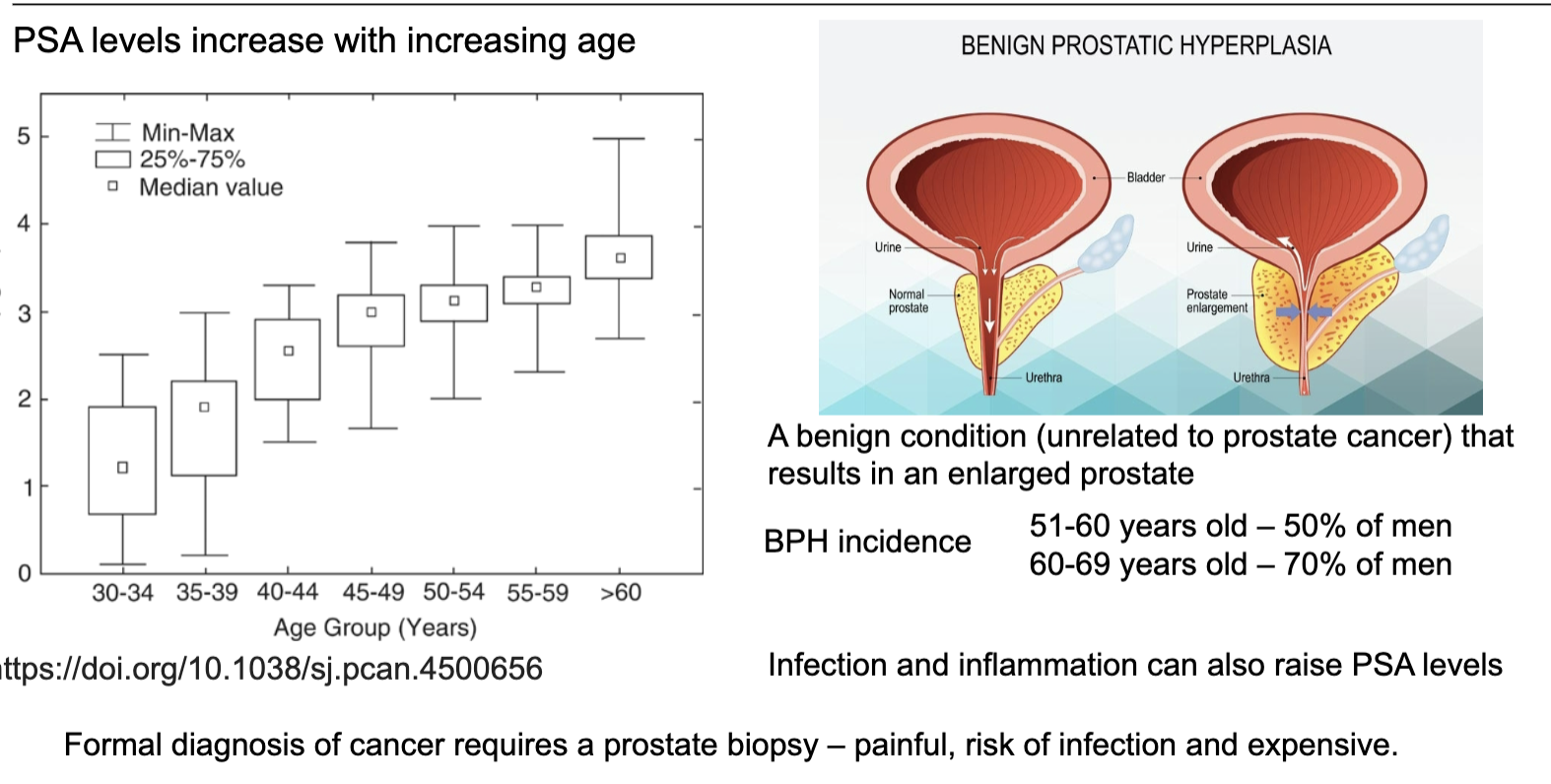
what is PSA
Prostate specific antigen (PSA) – a molecule expressed by prostate cells (normal and cancerous) detectable in the bloodstream
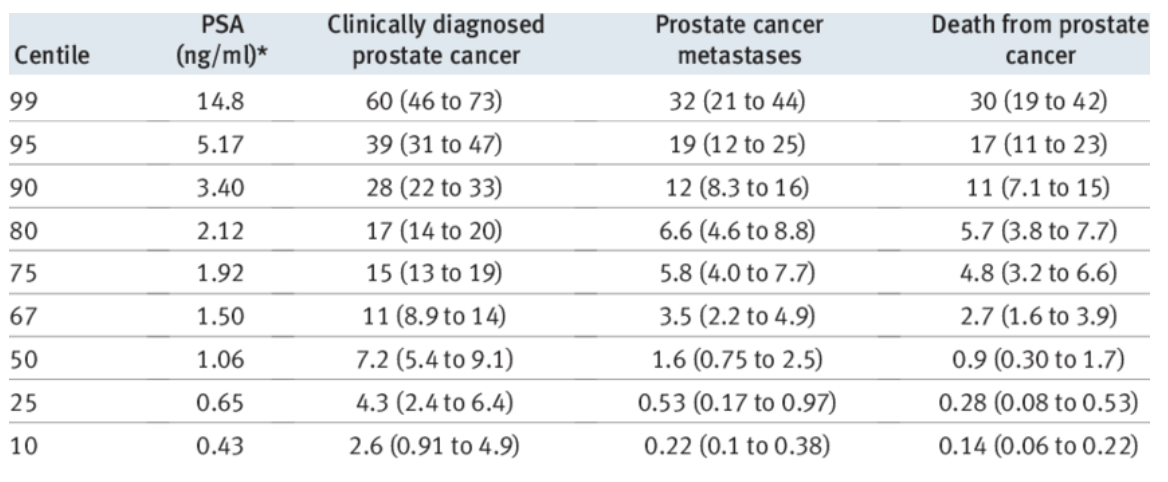
How does PSA level at age 60 relate to future prostate cancer risk?
Higher PSA at age 60 → higher lifetime risk of prostate cancer, metastasis, and death
Risk increases in a stepwise fashion across PSA percentiles
Even moderately raised PSA significantly increases long-term risk
However, PSA testing still has high false positive and false negative rates
Therefore, PSA is useful for risk stratification, but not a perfect diagnostic test
do all people with prostate cancer die from it/have the same prognosis?
no, the outcome of prostate cancer patients very heterogeneous
Only 10-15% of prostate cancers are aggressive (fast growing, more likely to spread)
85-90% are indolent (slow growing, less likely to spread)
most men die with prostate cancer than because of it
what is needed to account for prostate cancer outcomes being very heterogeneous
We need better diagnostic markers for differentiating between aggressive cancers that kill patients and those that are less aggressive and are not life limiting
Test results also need to indicate a clear clinical intervention (ie. help doctors and patients to make decisions on the way forward)
what is the natural history of chronic lymphocytic leukaemia (CLL)
clinical course of disease is very heterogeneous
monoclonal B cell lymphocytosis (asymptomatic)
CLL diagnosis (asymptomatic or weakly symptomatic)
treatment (heavily symptomatic)
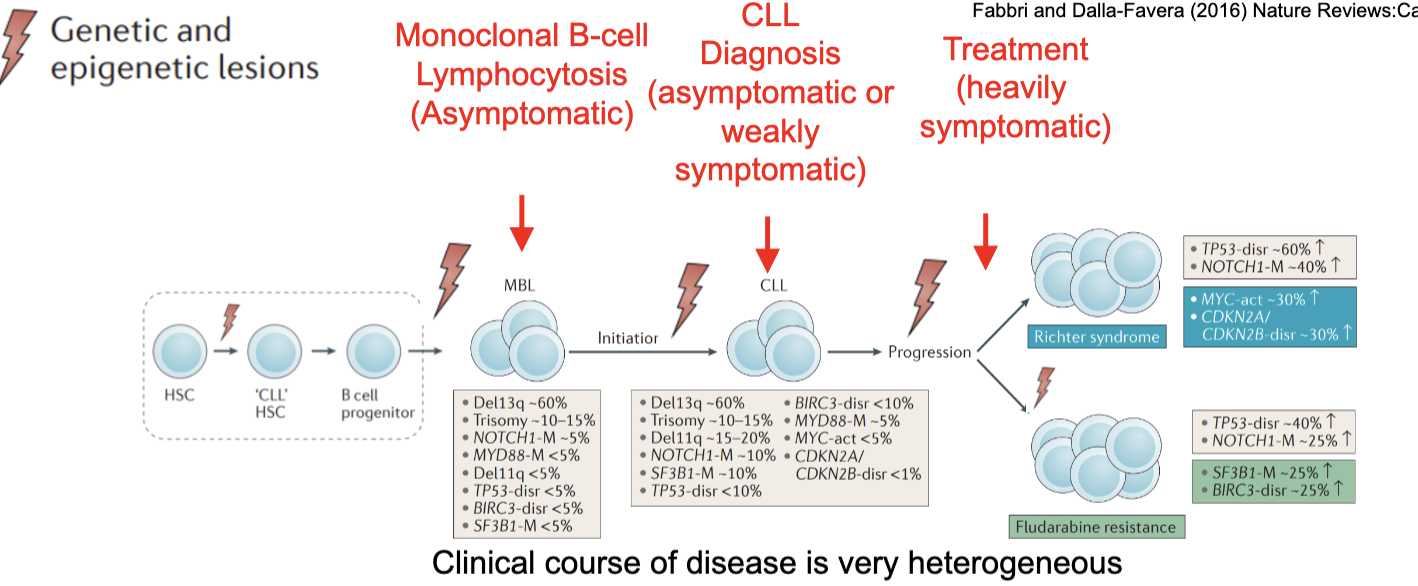
describe the outcome of patients with CLL
Most patients are diagnosed with early stage A disease and many will never require treatment (dying with CLL, not because of it) but approximately 40% will progress to requiring treatment.
Clinical strategy for stage A patients is currently “watch-and-wait”
is there a benefit to treating CLL early
Ibrutinib - Bruton’s tyrosine kinase inhibitor
Need to accurately identify those patients who will develop symptomatic disease and need treatment.
Don’t want to treat patients who will never progress
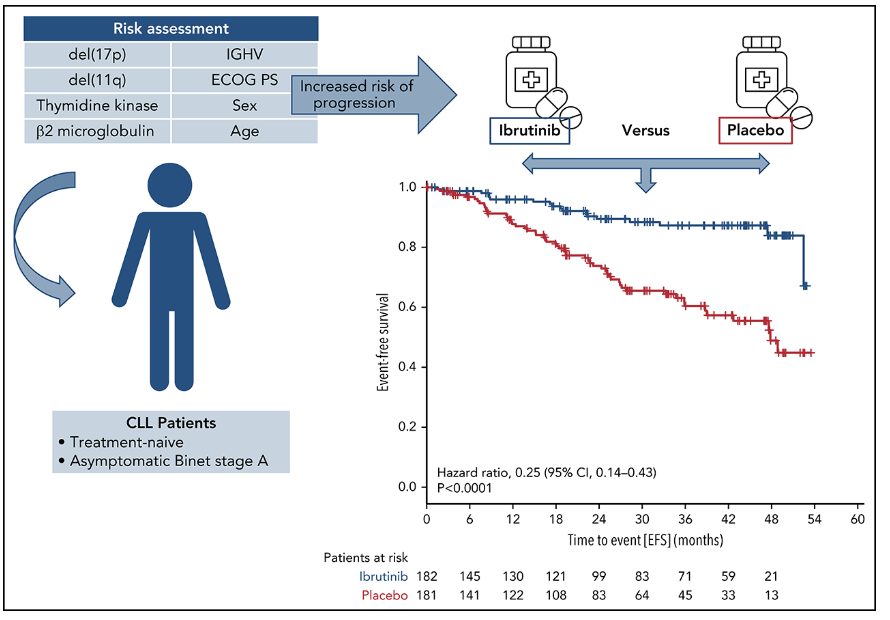
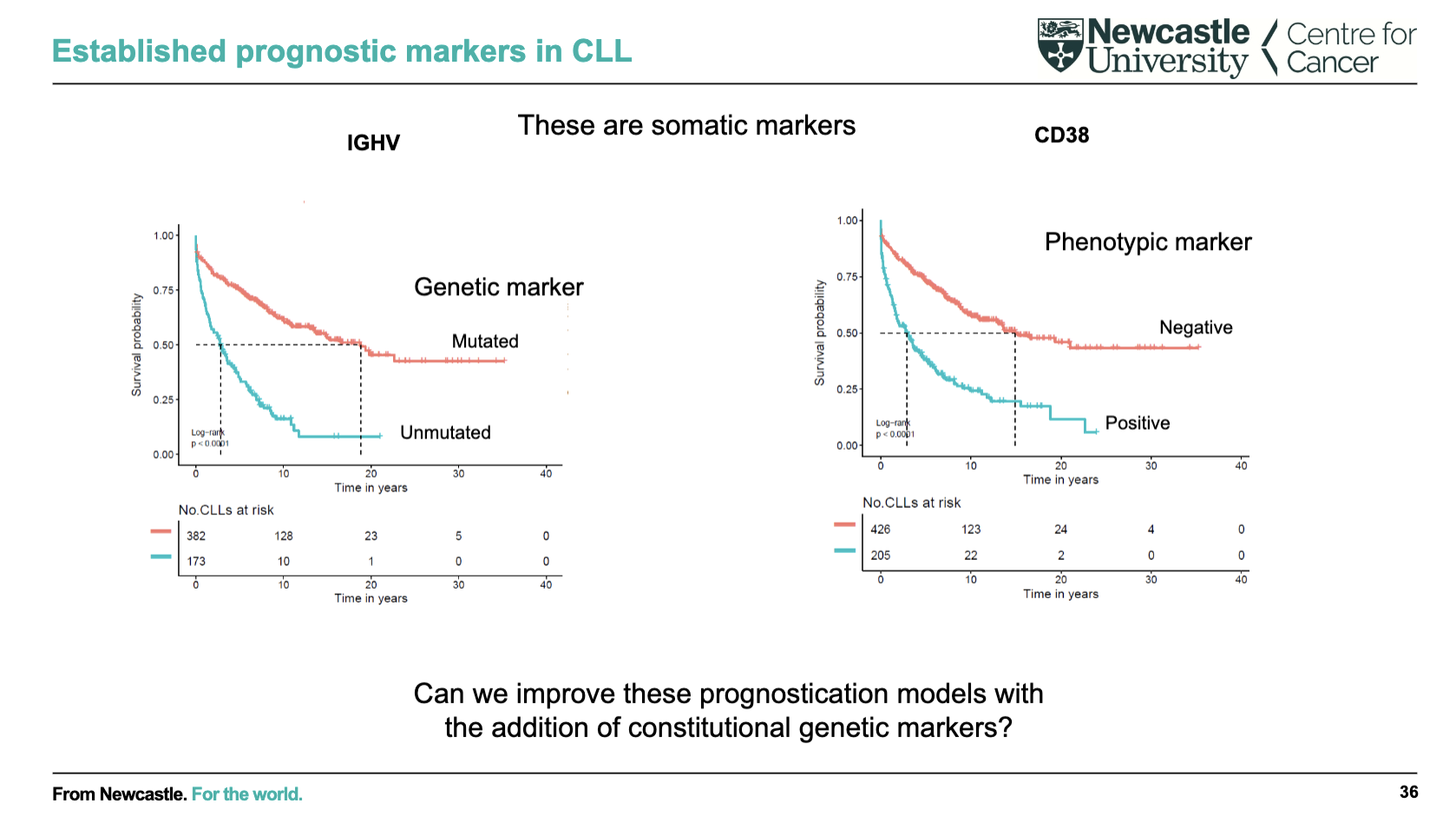
What are key prognostic markers in CLL and what do they indicate?
IGHV mutation status (genetic marker):
Mutated → better prognosis (longer survival)
Unmutated → worse prognosis
CD38 expression (phenotypic marker):
Negative → better prognosis
Positive → worse prognosis
These markers help predict disease course and survival
Ongoing question: can adding constitutional (inherited) genetic markers further improve prognostic models?
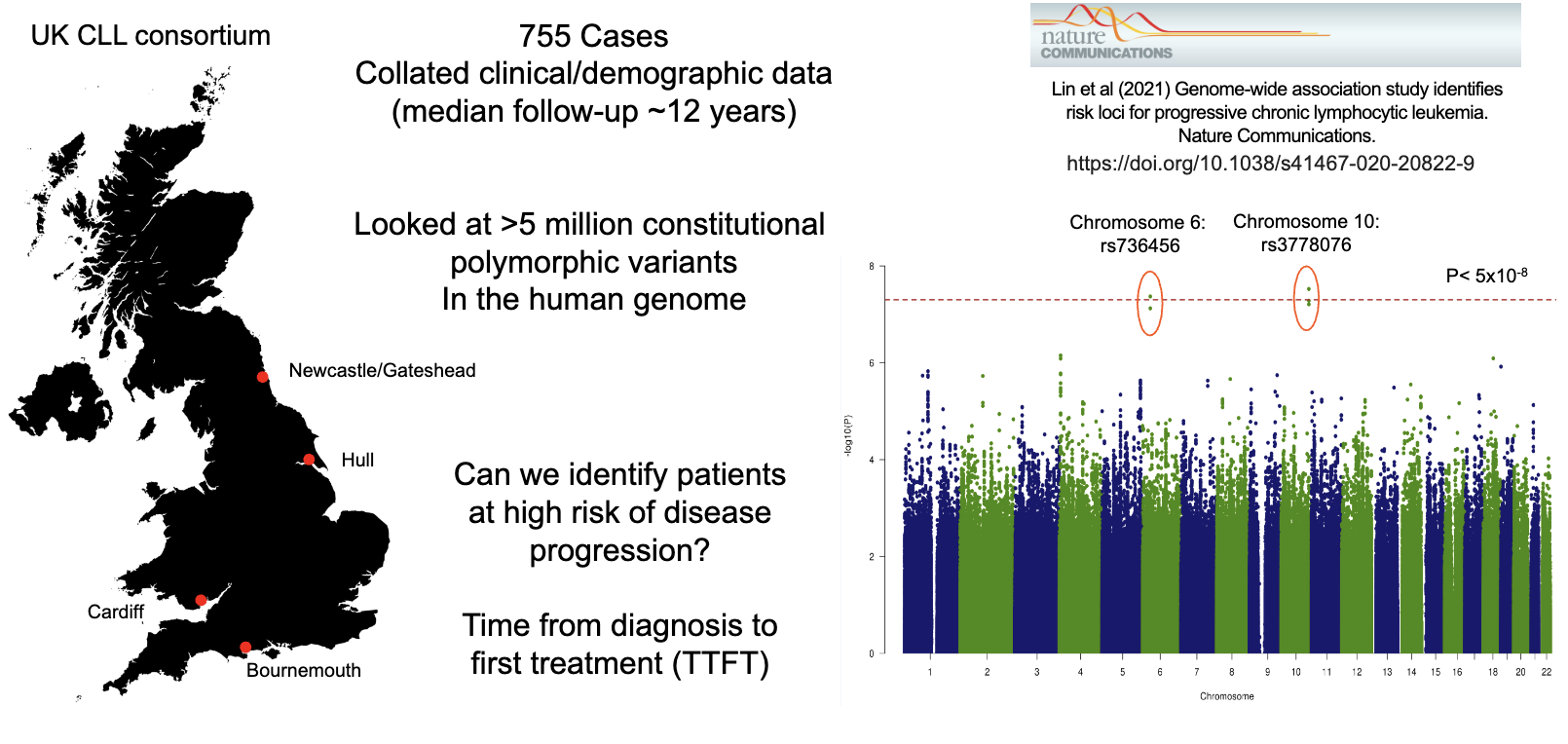
What is the aim of prognostic genome-wide association studies (GWAS) in CLL?
Analyse millions of genetic variants across patients to find risk-associated loci
Link inherited (constitutional) genetics with disease progression risk
Identify patients at high risk of needing earlier treatment (shorter TTFT)
Large datasets (e.g., UK CLL cohort) improve reliability of findings
Goal: enhance prognostic models beyond existing markers
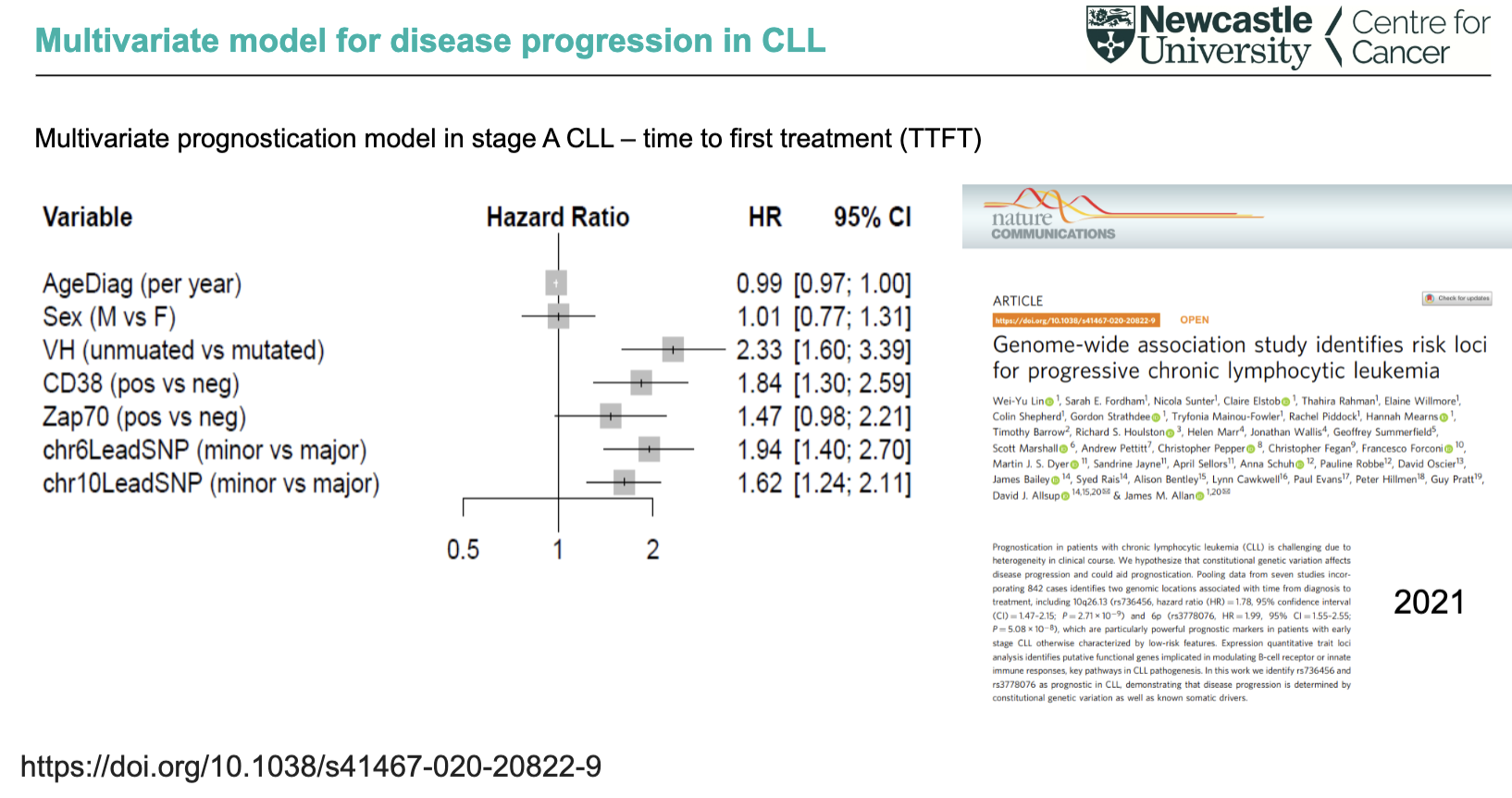
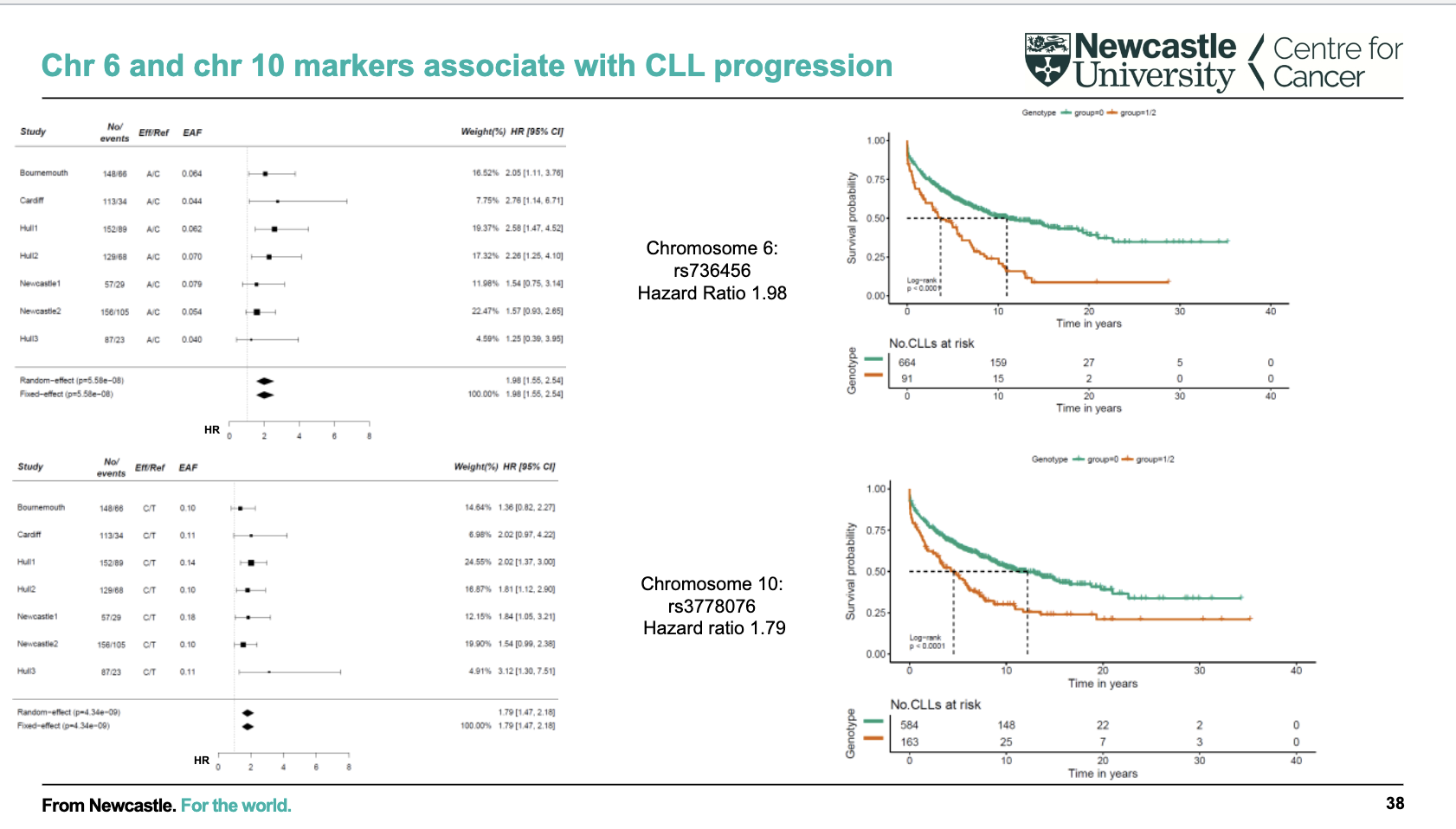
What is the significance of chromosome 6 and 10 markers in CLL progression?
Specific genetic variants on chr6 (rs736456) and chr10 (rs3778076) are linked to CLL progression
These variants are associated with shorter survival / earlier need for treatment (worse prognosis)
Effect size is moderate (hazard ratios ~1.8–2)
Findings are consistent across multiple cohorts (meta-analysis)
Supports use of inherited genetic markers to improve prognostic prediction
What does the multivariate model show about predictors of CLL progression (TTFT)?
Strong predictors of faster progression:
Unmutated IGHV (worst prognosis)
CD38 positive
chr6 and chr10 genetic variants
ZAP-70 shows weaker/borderline significance
Age and sex have little to no impact
Combining clinical + somatic + inherited genetic markers improves prediction of time to first treatment