protein purification and analysis
1/25
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
26 Terms
why do we need to purify proteins?
To separate them from the thousands of
other cell components to study their:
• Properties
• Activity
• Interactions with other molecules
• Structure
• Function
where do we get proteins from?
highly abundant in tissues of the body and then can be easily purified
Histones: from turkey blood (practical 4)
Haemoglobin: also from blood
Actin: from muscle
Tissue is obtained from animals at abattoirs (pig, cow, chicken etc.)
how do we generate enough protein for those that have low abundance?
Use a bacterial expression vector
- Strong promoter drives high protein expression in the cell
- Rapid replication of bacteria allows production of large quantities
How can we purify the protein from other cell
components so that we can study it?
Exploit the fact that each protein has unique
• Size
• Charge
• Shape
• Binding properties
how can we purify the protein from other cell components so that we can study it?
Several chromatographic methods allow us to do this, including:
• Ion-exchange separates on basis of charge
• Gel filtration separates on basis of size
• Affinity separates on basis of unique binding properties
first step in purifying proteins- get the proteins out of the cells
if its a muscle you freeze it in liquid nitrogen then crush it to break it up
first lyse the cells to obtain a cell extract
• If using tissue: mince it up first
• Use a detergent (e.g. NP40) to lyse (break open) the cells and release the contents
• Centrifuge the sample to separate out the part of the cell required (nuclei, mitochondria, cytoplasm etc.)
• Ultimately the protein of interest should be found in the liquid supernatant after centrifugation
(supernatant – Latin, meaning ‘floating above’)
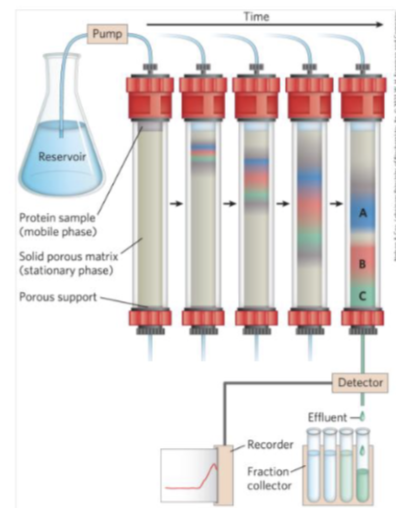
second step- basics of column chromotograogy
supernatant pass through the column - over time different proteins will separate out differently due to size and charge (according to properties of column that’s being used
eg in C you have to collect several drops and then protein B comes out and drips into the next tube- allows the proteins to be separated
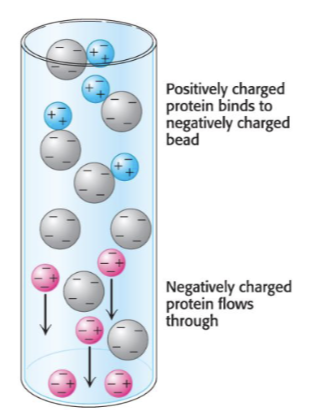
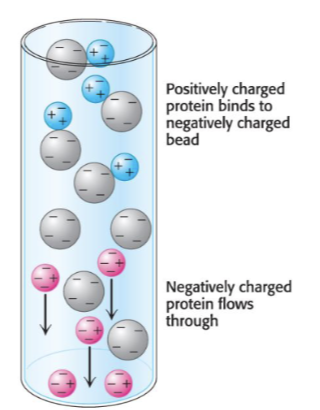
another way to purity - ion-exhancge chcomorograohy
Separation of proteins based on their charge
• The matrix is charged (either negative or positive)
• Bound protein can be eluted (released) by
changing concentration of sodium chloride
extra:
any positive will stick to beads and anything neg will pass through and won’t stick so will eliminate negatively charged proteins as it will repel from meg charged matrix , the positive charged will stick and elute off later by changing the conc of NaCl
another method: gel filteration
separate based upon size- large proteins can’t enter the mesh work so they pass through quickly and small proteins get trapped inside the matrix and then take longer
Separation of proteins
based on their size
• Small proteins move
slowly through the gel
column
• Large proteins move
quickly through the
column
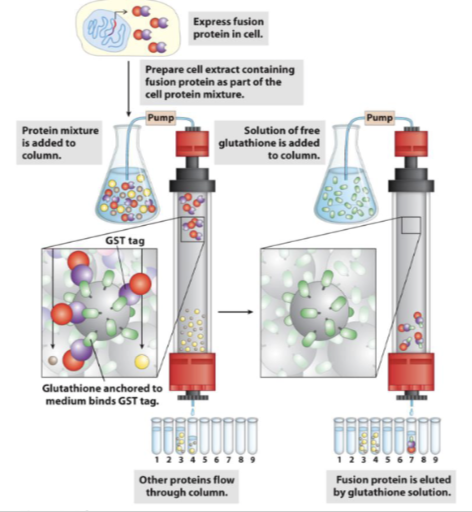
affinity chromatography
artificially engineer protein to have a tag on it so we can identify it
A unique protein sequence (affinity tag) usually fused to N- or
C-terminus of expressed proteins to allow purification
• Gene encoding target protein is fused to the gene coding for
the affinity tag e.g. glutathione-S-transferase (GST)- catalyses the reaction - binds glutathione so generate a column based on binding to separate out the protein
This will produce a GST fusion protein (first single messenger RNA is produced and then makes this protein)

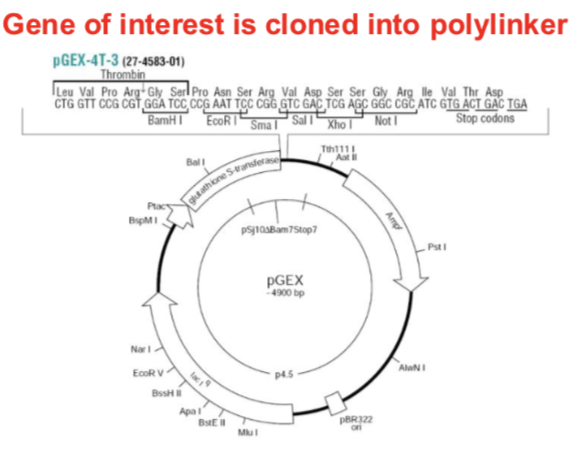
pGEX expression plasmids
• GST gene upstream of polylinker
• Bacterial promoter (Plac)
• RNA polymerase transcribes to
produce a single mRNA
• Produces large quantities of GST
fusion protein
Gene of interest is cloned into polylinker
affinity chromotograohy - AFTER card 12 - GST tagged proteins
GST fusion proteins can bind to an affinity
resin e.g. glutathione-agarose
Protein can be eluted from the affinity resin by
addition of a ligand which will compete for
binding to the resin e.g. free glutathione
so then only that protein will stick to the column
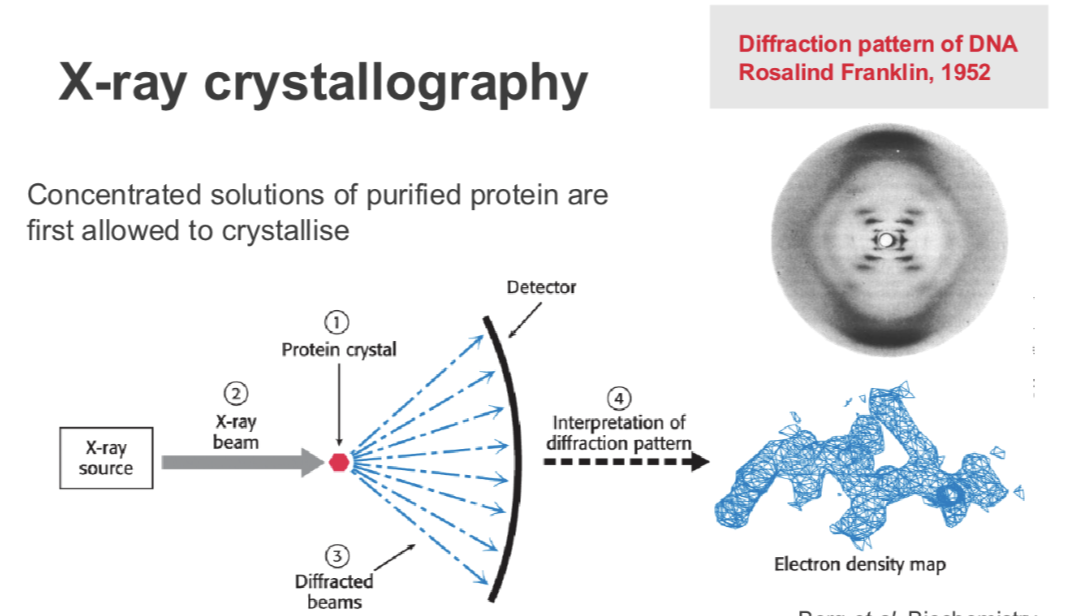
what is X-ray crystallography
Concentrated solutions of purified protein are first allowed to crystallise
direct an x-ray beam to the crystals and the
what does atomic structure allow us to understand
how proteins and molecules function
• The 3-D organisation of amino acids
• How enzyme active sites are structured
• How proteins interact with each other- eg with other proteins and substrates
Atomic structure allows us to understand
disease and design drugs to treat them
example in atomic structure understanding
BRAF- enzyme that’s commonly mutated in cancers
V600E mutation is most common
• Changes valine (V) to glutamate (E) at residue 600 of the protein
• How does this affect protein structure?
• The active site forms a hydrophobic pocket
• All the amino acid residues lining the pocket
have hydrophobic side chains
• Introduction of the negatively charged
glutamate changes the properties and the
shape of the pocket
• Switches the protein to its active conformation
➢Solving the structure has shown that it switches the protein to its active conformation
a drug that binds specifically to the mutant form of the protein in inhibit its function
• Zelboraf designed to fit into the
BRAF enzyme active site
• Blocks its function
• Prevents cancer cells from
proliferating
• Used to treat melanoma
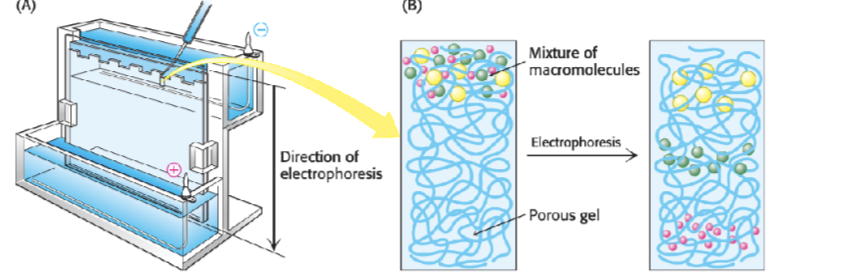
what is SDS=PAGE gel electrophoresis
Sodium dodecyl sulphate polyacrylamide - a way to separate proteins based on their size, smaller proteins will run through the matrix more quickly whereas large proteins will struggle to travel and go less far
Analogous to agarose gel electrophoresis of DNA
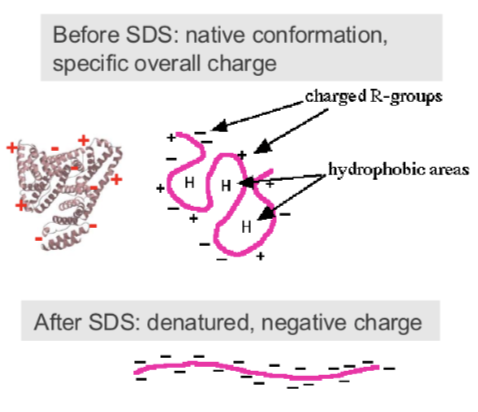
theory of SDS-PAGE
Separates proteins according to their size
• Under native conditions each protein has a unique shape, charge and size
• SDS is an anionic detergent that:
- denatures (unfolds) to uniform globular shape - due to electrostatic bonds breaking
- adds consistent negative charge- bigger proteins will have more negative charge (remember it wants to move to the positive side)
Proteins then separate on the gel purely according to their size (molecular mass)
how to visulise and detect proteins
using dyes or antibodies
how can we detect SDS-PAGE gel be directly detect using chemical dyes
proteins less abundant will have a weaker band, • These dyes bind to all proteins equally
• Hence they are not selective for individual proteins
• But they do reveal differences in the amounts of proteins
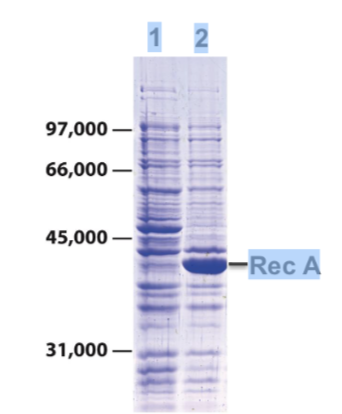
how interpret different lanes
• Lane 1: bacterial cell lysate separated by SDS-
PAGE
• Stain gel with Coomassie blue
• Reveals hundreds of different bacterial proteins
of different sizes and abundance
• Lane 2: lysate from cells expressing the protein
Rec A from a bacterial expression plasmid
BUT: many proteins are not expressed at
high enough levels to be visible
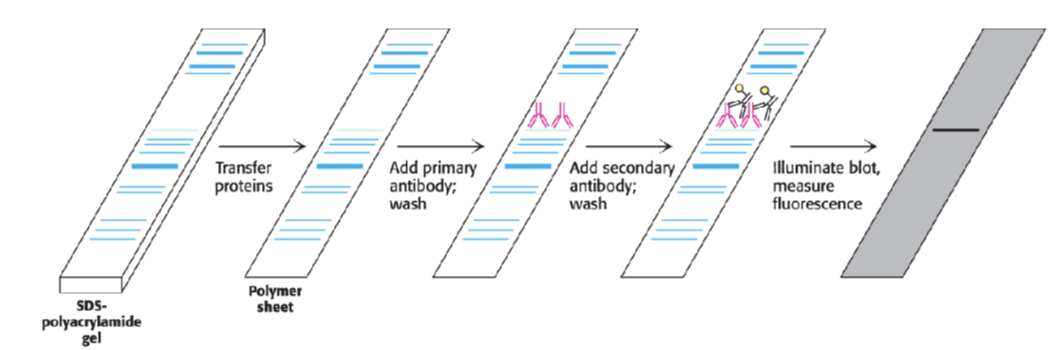
what can we do if proteins aren’t visible
use antibodies ,
transfer onto a membrane with antibodies which are designed to bind to something specific
how can we study proteins inside cells
• Immunofluorescence microscopy
• Expression of tagged proteins (fusion protein)
how does GFP work to visualise proteins in cells with a fluorescent tag
• GFP is derived from jellyfish
• Gene was isolated and cloned into plasmids
• GFP-fusion proteins are generated using recombinant plasmids
allows us to look in live cells
occurs naturally
can view throughout cells
protein tagging doesn’t fix the cell so it moves around
immuno kills the cell so you can only see in the moment so if you want to see it live you cant