BMM Module 12: Molecular Docking Simulations
1/21
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
22 Terms
Docking simulations
Simulation of binding between small molecule and a protein, where the output is a set of protein-molecule complexes with assigned scores
Docking simulations differ by
Scoring function
Force field based
Emperical
Knowledge based
Algorithm
Deterministic = predefined procedure with reproducible outcome
pros: fast and reproducible outcom
cons: may miss solutions
examples
Brute force
Shape fitting
incremental construction
Stochastic: includes randomness so different experiments can give different results
pros: better exploration of search space
examples
genetic docking
Monte Carlo
Tabu list search
Small molecule docking:
1 protein vs 1 ligand (class)
To understand how a ligand binds
Pose = different ways for a ligand to bind to a pocket
method works best when you use several similar ligands whose binding is known
Docking all creates many poses that can be clustered to identify common binding modes
Analyze
is there a correlation between the score and the experimental affinity
Do differences in bindingmodes explain differences in affinity
Small molecule docking:
1 protein vs many different ligands
For drug discovery
virtual screen of 1k to 10M compounds to identify which will bind and select those with the highest predicted affinity
Challenges:
as the amount of ligands increases, the score distribution widens and the top scores aren’t always active and you might miss hits
Solution: Consensus scoring = combine multiple scoring functions and select those that score well in all of themDifferent ligands might prefer different poses and it might be hard to select the best ones
some scoring function might give high score to pose that is not biologically relevent
Solution: combine docking with other methods (MIFs, Pharmacophore modelling, similarity to other compounds)
Incremental construction algorithm
Deterministic: fast and reproducible
fragment the ligand
choose core fragment (largest/most rigid)
Place core fragment in pocket and try all different orientations using shape complementarity
Incremently (1 by 1) add remaining fragment back to their original positions trying different torsions around the bonds
at each step only conformations that fit and avoid steric clashes are kept —> tree diagram
when fully reassembled: you have a set of ligand poses that can be scored
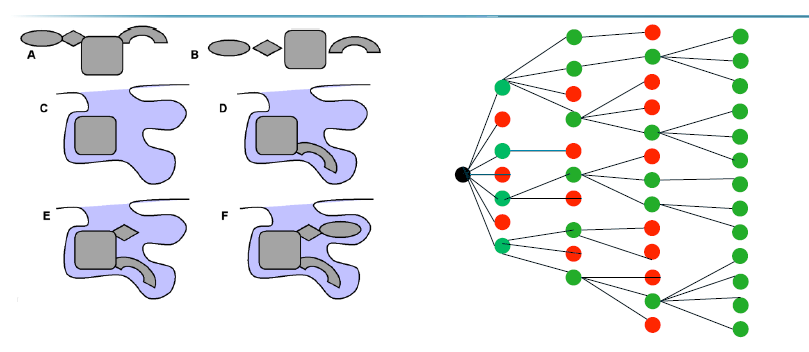
Genetic docking algorithm
Stochastic: evolutionary principles with random variations
Treat poses as a population of organisms that evolve over time
Each pose (1 solution) is encoded as a chromosome containing genes that encode information like atom coordinates, orientation, torsion angles
Start with a random population of chromosomes (diverse ligand poses)
Evaluation:evaluate fitness of each pose with fitness score
bad ones die
good ones survive
all poses where ligand binds outside of active site die, only the ones that bind in active site survive and reproduce
Reproduction: surviving chromosomes can reproduce via:
combining parts of 2 good solutions = crossover
randomly changing genes = local search
Repeat evaluation and reprodcution for many generations
Stop when:
top solutions don’t change significantly (RMS difference)
preset number of generations is reached
Similarities and differences Monte Carlo and Simulated annealing
Similarities
Both are stochastic methods (use random sampling to explore the search space)
Both used in Sampling of the PES to find low energy protein folds and in docking to find low energy protein ligand poses
Iterative processes: both generate and evaluate multiple candidate solutions over time
Differences
Goal
GA: find the best possible solution (optimization)
output = best found solution
MC: random sampling
output = distribution of potential outcomes
Approach:
GA works on populations of solutions
MC works on a single solution at a time
Mechanism
GA: solutions can share information via crossover
MC: No information exchange between samples
Evaluation
GA: uses score to compare and rank multiple solutions within a population (determines which survive and reproduce)
MC: uses scoring function to probabilistically accept or reject a move relative to the current state (not to rank solutions)
other algorithms
stochastic
monte carlo
tabu list search
deterministic
brute force
shape fitting
Tabu list search
several initial ligand placements are generated randomly near the bindning site
to have multiple starting points but only one pose per run not multiple at the same time
The molecule is moved by small changes
Each new position is scored
moves or positions that appear in the Tabu List are not allowed
move selection
better-scoring moves are preferred
worse moves may be chosen if no better non tabu move is available (to escape local minima)
memory update
the selected move is added to the tabu list
so they’re temporarily forbidden to prevent revisiting them
steps 2-6 are repeated until convergence
Brute force
try every possible orientation/postion of the ligands
shape fitting
generate a low energy conformation of the ligand
fit the conformation in the pocket based on geometric constraints
optimize and score the fit
often used for fast screening
Scoring function
Biophyisical formulas that describe the quality of a docked molecule to its receptor
quality score during docking to guide algorithm towards better poses
quantitative score to rank docked molecules according to binding strength after docking
Force Field based scoring function
Only take non bonded interactions between protein and ligand into account (gives energetic values)
cons:
no entropic contribution
no inclusion of water models
water can mediate key ligand-receptor interactions (H-bonds)
FF parameters are hard to parameterize for a specific target
FF rely on param like partial charges, atom types, … which are general and not tailored for a specific protein so the FF may not describe interactions accurately for a particular target
Emperical scoring function
= estimates protein-ligand binding by summing weighted interaction terms (H-bonds, hydrophobic,…) where the weights are obtained by fitting experimental binding data of known protein-ligand complexes using regressionmethods (=adjusting weights to match exp data)
Free energy terms
\Delta Gx =contains all unknown contributions learned through regression
polar interactions: H-bonds and ionic
apolar interactions: aromatic and lipophylic
entropic effects:
desolvation effects (removal of water upon binding)
loss of ligand flexibility: as ligand binds, the number of rotable bonds decreases ~Nrot
cons
need a training set
scores are only good if problem resembles training set
Weights=\sum f\left(\Delta R,\Delta\alpha\right)
\Delta R,\Delta\alpha —> The more you deviate from the ideal fitted angle/distance the worse the score will be and how much worse is determined by the weights
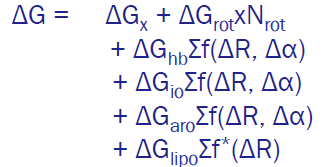
Knowledge based
uses statistics from known crystal structures to determine how favorable an interaction is
Get stratistics from protein data bank: how often does atomtype i interact with atomtype j at a certain distance
build histograms of distance vs frequency of interaction
convert frequency to energy (more common interactions = lower energy)
apply a scaling factor: some atomtypes occur more in crystalstructure than others
Resulting score ranks the quality of the predicted pose (not true energies)
Rosetta score is a scoring function for folding and design and is a knowledge based scoring function
Challenges for scoring functions
water
Water kan form upto 4 H-bonds and a lot of protein ligand interactions are mediated by water
but this is hard to model
In general, important water form >2 H-bonds
Induced fit
Most docking software keep protein rigid while flexible ligands are docked, but in reality the protein can change conformation as a ligand binds = induced fit
Solution:
Allow rotation of polar H’s or alternative rotamers (= alternative side chain conformations)
Energy mininmize after docking
run MD for realistic flexibility
Over and underscoring
scoring functions are additive so more interactions means a higher score
Big molecules in general have higher scores because they have more contactpoints
So it’s hard to compare molecules of different sizes
Docking produces many possible poses
false positives= bad pose with high score
false negatives = good pose with low score
solution: consensus scoring: combine multiple scoring functions
solvation and desolvation is not taken into account
Advanced fix to scoring function challenges
Use MM-PBSA after scoring:
inlcudes solvent
accounts for some induce fit through MD simulations
less dependent on the amount of heavy atoms in the ligand (less overscoring)
inludes entropy - desolvation effects (energetic consequences for removing water from both the ligand and receptor when they bind)
MM-PBSA: concept
= post processing method use to estimate the binding free energy of a ligand to a receptor more accurately than simple scoring functions
MM = molecular mechanics: calculates internal energies of ligand receptor and complex
bonded+non-bonded terms
PBSA = poisson boltzmann surface area
estimates 2 solvation effectspolar solvation = electrostact stabilization by water using the poisson boltzmann formula
apolar solvation = energetic penalty of creating a cavity in water (related to solvent accessible surface area)
the binding free energy is computed
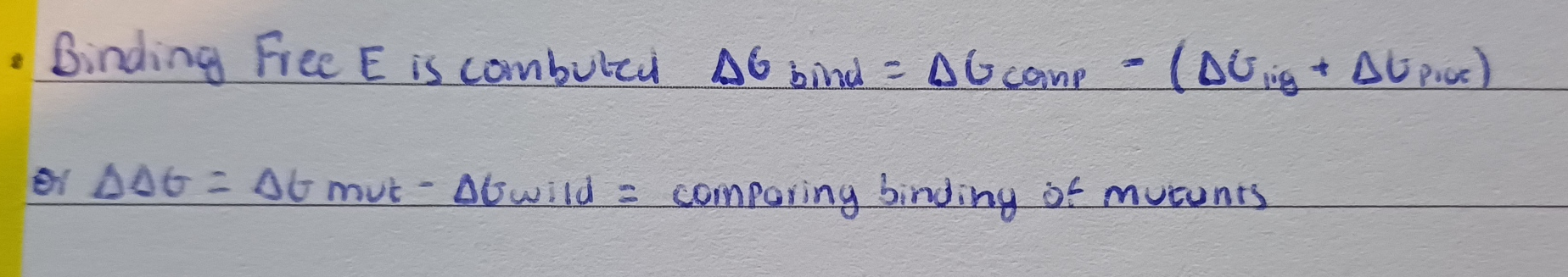
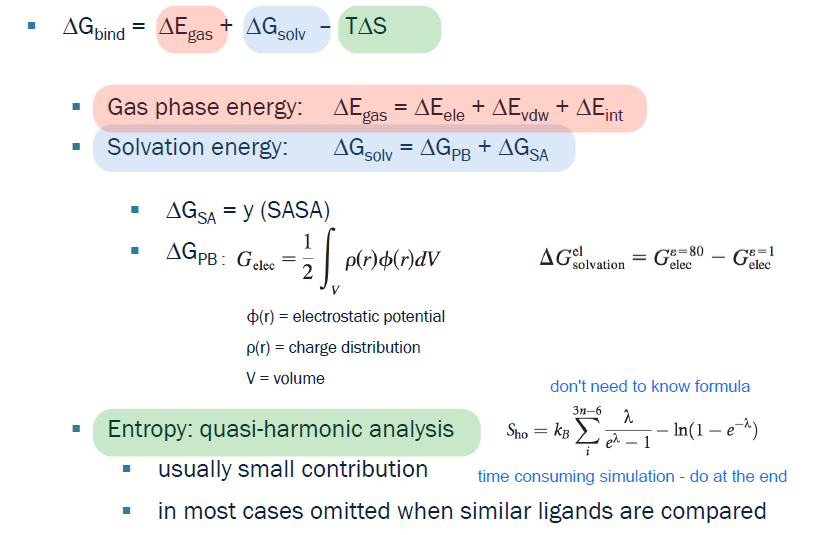
MM/PBSA workflow
run short MD (10ns) of the protein ligand complex with GROMACS to sample realistic conformations
extract snapshots
compute the energy of the complex, protein alone and ligand alone
compute the MM gas phase interaction energies: electrostatic VDW and bonded terms
Compute solvation effects with the implicit solvent model (MM-PBSA uses an implicit solvent model, even if the underlying MD simulation was run with explicit water.)
polar contribution: PB
apol contribution: SA
Calculate the binding free energy
optionally add an entropic term
Averaging
Average all binding free energies over all snapshots to obtain the final estimate
the more negative the binding free energy the stronger the binding
Analysis of your method
Before you use a method you want to analyze if it works, this is only possible if you know which comounds are active and inactive
This is done by using the method to dock compounds and rank them according to score ( the method should be better than a random selection of compounds)
Commonly used methods
ROC curve
enrichment factor
(but no perfect analytical method exists)
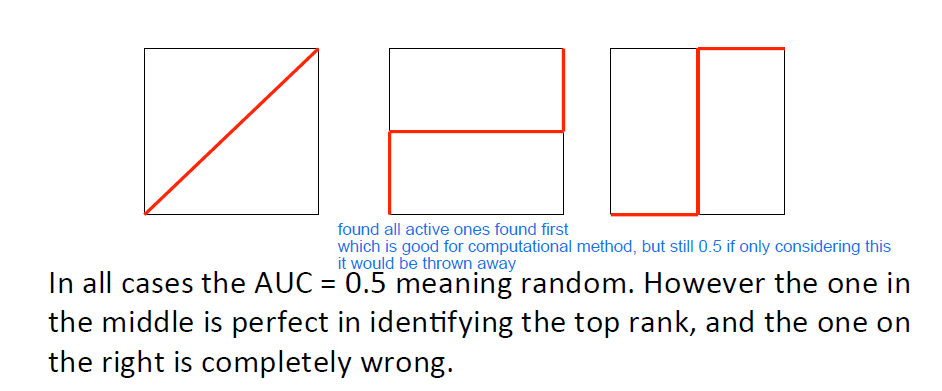
ROC curve
= receiver operator characteristic
plots true positive rate (TPR) vs false positive rate (FPR)
rank all docked compounds according to score
Imagine going down the list
if the first compound is a known active move up 1/A with A = # active compounds
If the next compound is a known incactive: move to the right 1/I with I = #inactive compounds
A perfect method ranks all active compounds before the inactives:
Area under the curve (AUC)
AUC = 1 for perfect method
AUC = 0.5 for random picking
AUC <0.5 worse than random
AUC is no sufficient as a metric therefore the enrichment can be calculated
Enrichment
Fraction of found actives in the hit database (obtained by docking method) compared to the fraction of actives in the whole database
