Cancer Genetics
1/51
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
52 Terms
Cancer
a group of diseases characterized by ______ (controllable/uncontrollable) growth and spread of ______ (normal/abnormal) cells
______ multiplication/_______ leads to the formation of a lump of tissue or tumor
tumor cells invade the __________ tissues and may travel to a distant site to form new tumors
the process by which tumor cells spread to distant sites is called ______
uncontrollable, abnormal
uncontrolled, proliferation
surrounding
metastasis
Cancer Risk Factors
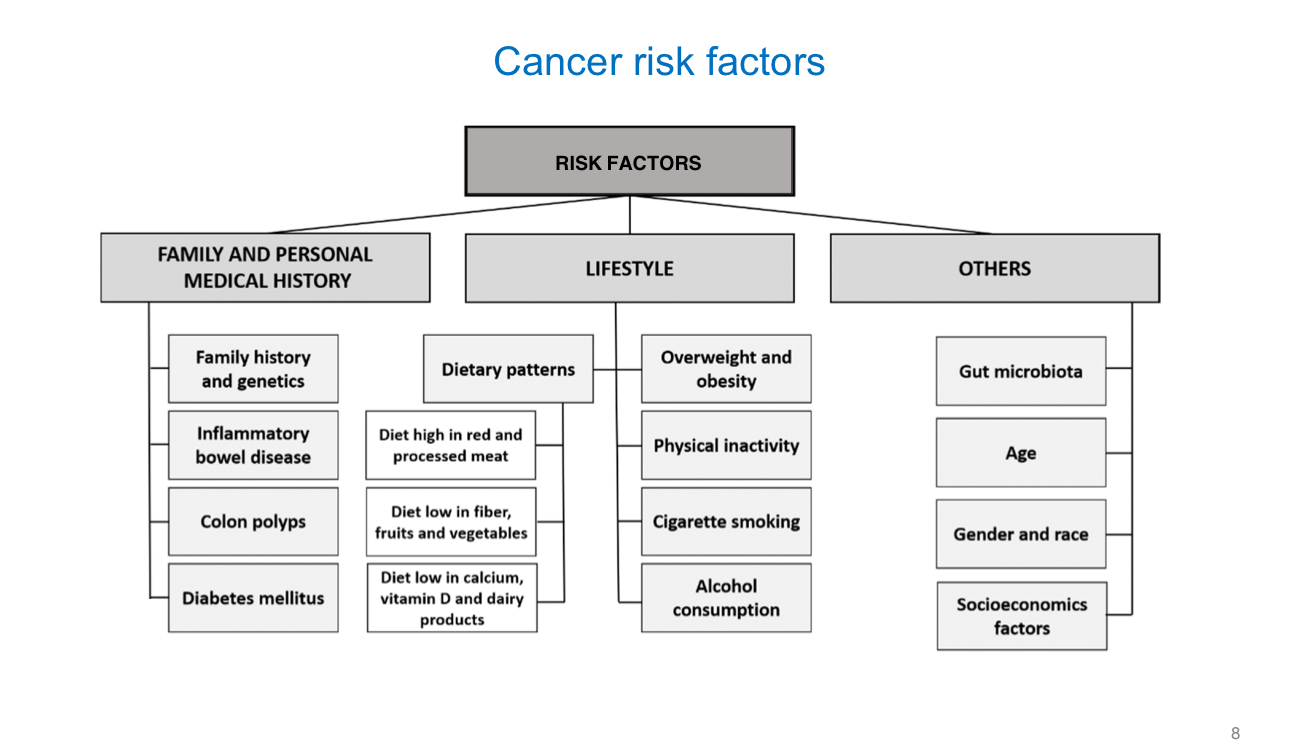
Carcinogens —> substances capable of causing cancer
Lifestyle Factors —> ______, ______, ______ light, ______ fat diet
Environmental Factors —> ______ radiation + ______ radiation
Chemical Agents —> ______ drugs, certain ______ agents, ______ ______
Infectious Agents —> (4)
smoking, alcohol, UV, high
UV, ionizing
immunosuppressive, antineoplastic, coal tar
hepatitis B/C, HIV, HPV, H. pylori
Etiology of Cancer
not fully ______
carcinogenesis is a ______ process regulated by ______ —> ______ steps
initiation: exposure to ______ that causes DNA damage —> ______ mutation
promotion: growth of ______ cells —> pre-neoplastic lesion —> ______ (reversible/irreversible)
conversion: mutated cell becomes ______ occurs __________ years after prior stages —> ______ (reversible/irreversible)
progession: further ______ changes, tumor ______ into local tissues, distant ______ —> ______ (reversible/irreversible)
understood
multistep, genes, four
carcinogen, irreversible
mutated, reversible
cancerous, 5-20, irreversible
genetic, invasion, metastases, irreversible
Tumor Origin and Classification
tumors arise from any _____ type and may be named based on tissue type:
epithelial —> ______
connective (muscle, bone, cartilage) —> ______
lymphoid/blood/bone marrow —> ______, ______, ______
tumors may also be classified as:
______ site —> lung, colon, etc.
degree of ______ (poorly differentiated vs. well differentiated)
______ (insulinoma)
______ or ______
tumors that end in “-oma” —> ______
tumors that end in “-carcinoma” —> ______
a tumor is treated according to the site of ______
tissure
cacinoma
sarcoma
lymphoma, leukemia, myeloma
anatomic
differentiation
function
benign, malignant
benign
malignant
origination
Benign vs. Malignant Tumors
Benign
grow ______
______ capsule
______ (non-invasive/invasive)
______ (well/poorly) differentiated
______ (high/low) mitotic index
______ (does/does not) metastasize
Malignant
grow ______
______ encapsulated
______ (non-invasive/invasive)
______ (well/poorly) differentiated
______ (high/low) mitotic index
______ (does/does not) metastasize
slowly
well-defined
non-invasive
well
low
does not
rapidly
non
invasive
poorly
high
does
Benign Tumor Complications
Benign tumors can still cause complications such as:
__________
Brain tumors → __________ effects
Secretion of __________ (e.g., thyroid adenoma → thyroid hormone; pituitary adenoma → growth hormone)
Leiomyoma → __________ and __________ symptoms
pain
CNS
hormones
heavy menstrual bleeding, urinary
Cancer Diagnosis
requires a ______ (tissue sample) for pathological diagnosis
Types include:
______/______ biopsy
______ biopsy
______ cytology
biopsy
incisional/excisional
needle
exfoliative
Cancer Staging (TNM System)
staging is based on tumor ______, ______ ______ involvement, and ______
T (tumor):
Tx = tumor cannot be __________
Tis = carcinoma __________
T0 = no __________ of tumor
T1–T4 = size or extent of __________ tumor
N (node):
Nx = node cannot be __________
N0–N3 = degree of __________ to regional lymph nodes
M (metastasis):
M0 = ______ distant metastasis
M1 = __________ of distant metastasis
Stage I: ______ prognosis
Stage IV: ______ prognosis
size, lymph node, metastasis
evaluated
in situ
signs
primary
evaulated
spread
no
presence
best
worst
Cell Division and Cell Cycle
Interphase consists of three phases:
G₁ phase – gap between __________ and __________ phase
S phase – where __________ occurs
G₂ phase – gap between __________ and __________ phase
G₁ and G₂ allow __________.
M (mitotic), S (synthesis)
DNA replication
S, M
cell growth
How does cancer develop?
cancer is a ______ disease and ______ process that takes time to develop —> often years
it arises through a series of ______ alterations in DNA
a ______ mutation does not necessarily lead to cancer —> _____ mutations must also occur
usually, about ______ mutations are required for development of most cancers
at each step, mutated cells gain a __________ advantage, leading to __________ of those cells
the most important genetic alterations occur in __________ and __________
genetic, multi-step
somatic
germline, somatic
three
growth, expansion
tumor suppressor genes, proto-oncogenes
Germline vs. Somatic
Germline:
______ (inherited/acquired)
found in ______ and ______ cells and EVERY cell in the body
Somatic:
______ during a person’s lifetime
______ to specific cells/tissues
inherited
egg, sperm
acquired
limited
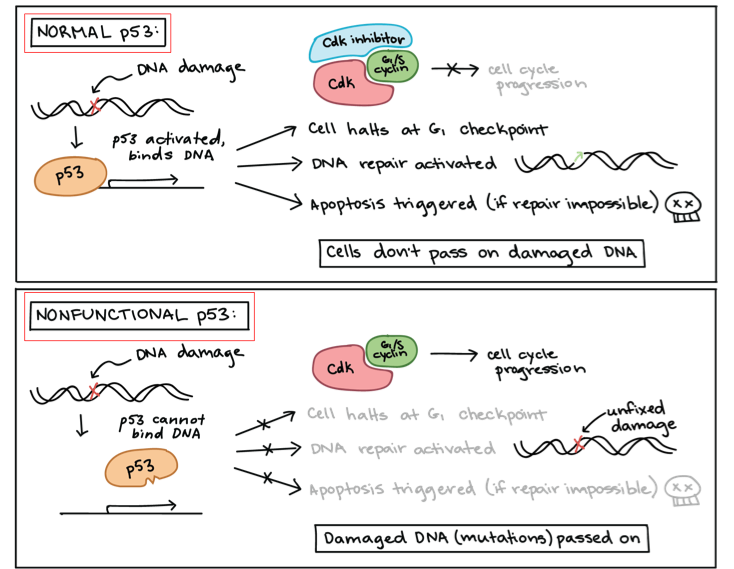
Tumor Suppressor Genes in Cancer Development
normally suppresses ______ ______, repair ______ mistakes or induce ______
inactivation of tumor suppressor genes can lead to ______ development
Mechanisms of Inactivation:
______ mutations
______/______
gene ______ —> epigenetic change
cell division, DNA, apoptosis
Tumor Suppressor Genes —> Examples
______
BRCA 1/2 → drug class: __________ inhibitors → example: __________
Retinoblastoma (Rb) → drug class: __________ inhibitors → example: __________
p53
PARP, olaparib
CDK4/6, palbociclib
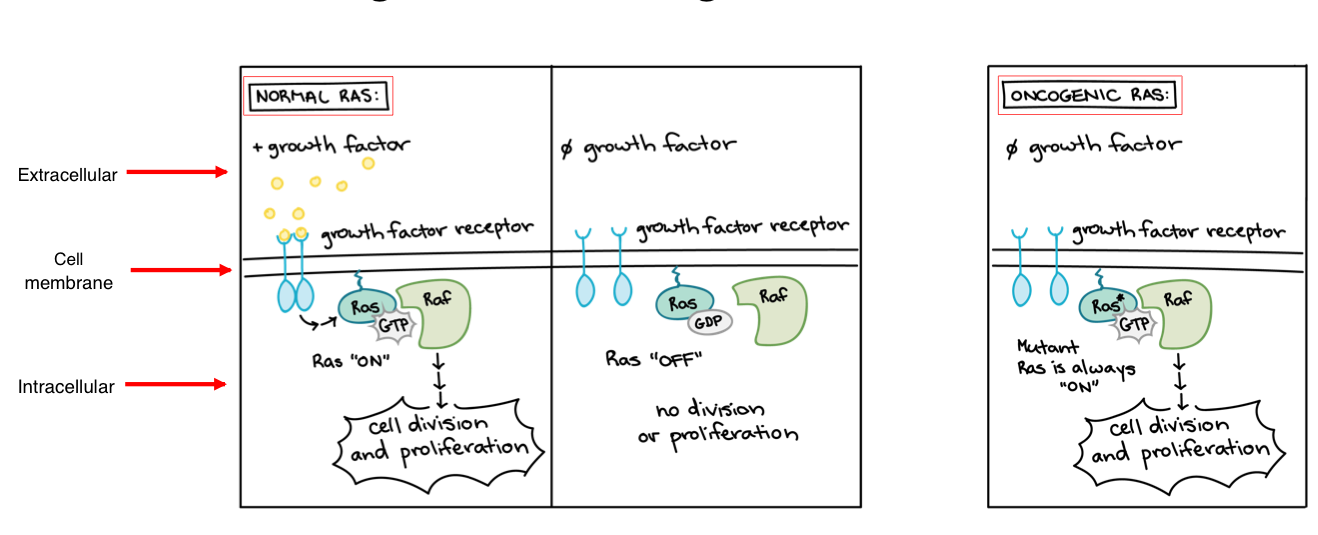
Porto-Oncogene in Cancer Development
promote ______ ______ or reduce occurrence of ______
when proto-oncogene mutates, they become ______ due to the over-activation of the gene
over-activation = ______ development
mechanisms of oncogene activation:
______ mutation —> RAS
DNA ______ —> HER2
chromosomal ______ —> BCR-ABL
cell division, apoptosis
oncogene
cancer
point
amplification
rearrangement
Oncogene Examples
RAS → inhibitor: __________
EGFR → inhibitor: __________
HER2 → inhibitor: __________
VEGF → inhibitor: __________
BCR-ABL → inhibitor: __________
sotorasib
osimertinib
trastuzumab
bevacizumab
imatinib
Retinoblastoma (Rb): Tumor Suppressor Genes
>70% of human tumors have a mutation that leads to the ______ or ______ of Rb
normally, Rb inhibits the ______ ______ by inhibiting transcription factors such as ______
CDK 2, 4, and 6 inactivate Rb via ______, releasing transcription factors that allow progression from ______ to ______ phase
relevant drug class —> ______ inhibitors —> ______ (Ibrance)
these drugs inhibit Rb ______/______, restoring Rb’s ability to suppress the cell cycle
used in the treatment of ______ cancer
loss, inactivation
cell cycle, E2F1-3
phosphorylation, G1, S
CDK 4/6, palbociclib
phosphorylation/inactivation
breast
BCRA1/BCRA2: Tumor Suppressor Genes
involved in the ______ ______ (HRR) repair pathway, which repairs ______ DNA (dsDNA) breaks
cells with BRCA1/2 mutations are deficient in their ability to repair dsDNA breaks and must rely on ______ repair mechanisms such as ______ ______ repair (BER)
BER is mediated by ______
relevant drug class: ______ inhibitors —> ______ (Lynparza)
PARP inhibitors block the alternative repair pathway used by ______ cells, leading to cell death through ______ ______
used in the treatment of ______, ______, ______ cancers
homologous recombination, double-stranded
alternative, base excision
PARP1
PARP, olaparib
BRCA-deficient, synthetic lethality
prostate, breast, ovarian
BCR-ABL: Oncogene
present in cells with translocation of chromosomes ______ and ______
the gene product (BCR-ABL fusion protein) has constitutive ______ ______ activity, which activates signaling pathways via ______ of substrates
relevant disease state: ______ ______ ______ (CML)
relevant drug class: ______ inhibitors —> ______ (Gleevec)
BCR-ABL inhibitors bind to the ______ domain and block the ______ of the substrate
, 22
tyrosine kinase, phosphorylation
chronic myelogenous leukemia
BCR-ABL, Imatinib
kinase, phosphorylation
what is the first drug to inhibit a gene product that is only found in cancers?
Imatinib (Gleevec)
Growth Factors: Oncogenes
Several mechanisms in oncogenesis:
______ of growth factors
______ ______ of growth factor receptors
______ of growth factor receptors
Example: ______ ______ growth factor (VEGF)
relevant drug class: ______ inhibitors —> ______ (Avastin and biosimilars)
MOAs:
bind the growth factor (VEGF A/B) —> ______
bind the VEGF receptor to block binding of VEGF —> ______
inhibit VEGF receptor’s intracellular kinase —>
VEGF targeting agents inhibit ______
overproduction
activating mutations
overexpression
vascular endothelial
VEGF, bevacizumab
bevacizumab
ramucirumab
sunitinib
angiogenesis
Relevant Mechanisms of Drug Resistance
Changes in target
Acquisition of ______ mutation in gene
Upregulation of ______
Change in the affected pathway
Use of ______/______ or enhanced DNA repair pathways
Develop new ______/______ to bypass effects of the drug
Epigenetic changes
Turn genes __________ that affect treatment response
Development/enhancement of ______ ______ mechanism
new
expression
alternative/enhanced
pathways/signals
on/off
drug efflux
Cancer is a heterogeneous disease
Tumors have unique genomic and phenotypic features that vary at multiple levels.
Genetic heterogeneity:
Cells within a single tumor are not genetically identical.
Mutations accumulate over time during tumor growth.
Many mutations are passenger mutations (not clinically significant).
Driver mutations promote tumor growth/progression and are often actionable/druggable.
Levels of Cancer Heterogeneity
Cancer heterogeneity occurs across three major levels:
Intratumor heterogeneity
Differences among cells within one tumor mass.
Multiple subclones exist inside a single tumor.
Intertumor heterogeneity
Differences between a patient’s primary tumor and their metastatic tumors.
Interpatient heterogeneity
Differences between tumors found in different patients.
Why is tumor heterogeneity important?
1. Interpatient heterogeneity
Different tumor types require different treatments (e.g., breast cancer vs prostate cancer).
Even tumors of the same type may need different treatments due to differences in each patient’s tumor genetics.
This concept forms the basis of precision medicine, where treatments are tailored to the specific molecular features of each patient’s cancer.
2. Intratumor and Intertumor heterogeneity
These types of heterogeneity can strongly influence drug response and resistance.
If all tumor cells shared the same genetic makeup, they would likely respond uniformly to therapy.
However, because tumors contain multiple genetically distinct subclones:
Some cells may respond to treatment.
Others may survive and lead to treatment failure or resistance.
This genetic diversity makes it less likely that a single therapy will eliminate all tumor cells.
Intratumor / Intertumor heterogeneity
The effectiveness of targeted therapy depends on whether the target mutation is present in all tumor cells.
Example: If a tumor has an EGFR mutation, an EGFR inhibitor can produce a complete response only if every tumor cell carries the mutation.
If some cells lack the mutation → they survive, leading to resistance and tumor regrowth.
Combining drugs with different mechanisms of action may help eliminate diverse tumor cell populations and reduce resistance.
Models of Tumor Heterogeneity
1. Clonal Evolution (CE) Model
All malignant cells start as biologically similar.
Over time, they accumulate genomic or epigenetic alterations in a stepwise manner.
Selective pressures cause certain clones to expand while others die out.
This creates genetically diverse subpopulations within the tumor.
2. Cancer Stem Cell (CSC) Model
Tumor contains a small subset of cancer stem cells (CSCs).
CSCs:
Have the ability to self-renew.
Generate the heterogeneous populations of cancer cells in the tumor.
Only CSCs drive tumor growth, progression, and recurrence.
3. Plasticity Model (unites both CE and CSC models)
Cancer cells can interconvert:
Differentiated cancer cells ↔ cancer stem–like cells.
This means tumors can shift between CE-like evolution and CSC-driven behavior.
Clinical Application of Tumor Heterogeneity Models
Clonal Evolution (CE) Model
If this model is correct:
Most cells in the tumor can proliferate, metastasize, and contribute to disease progression.
Therapeutic implication:
Treatments must aim to eliminate most or all tumor cells, since many cells have malignant potential.
Cancer Stem Cell (CSC) Model
If this model is correct:
Only a small subset of cancer stem cells (CSCs) drive tumor initiation, growth, and progression.
Therapeutic implication:
Therapies should specifically target CSCs, because eliminating them could prevent tumor recurrence and progression.
Therapeutic Options Are Driven by Tumor Heterogeneity
Treatment depends on both:
Tumor type
Presence of actionable genetic alterations
Examples:
Cancer Type | Common Treatment (no mutation identified) | Targeted Treatment (if mutation identified) |
|---|---|---|
Non–small cell lung cancer (NSCLC) | Platinum + Taxane | - PD-L1 > 50%: PD-1 inhibitor - EGFR mutation: EGFR inhibitor - ALK rearrangement: ALK inhibitor |
Breast cancer | Anthracycline + Alkylating agent | - HER2+: trastuzumab, pertuzumab, docetaxel - Hormone receptor+: antiestrogen + CDK4/6 inhibitor |
Take-home points
Treatments differ based on tumor type and tumor genetics.
When cancer recurs, new mutations may appear, so re-testing is often needed to guide therapy.
Precision / Personalized Medicine
What is it?
A major clinical application of tumor heterogeneity.
Precision medicine = tailoring prevention and treatment strategies to the specific characteristics of the patient and their tumor.
Factors include genes, environment, and lifestyle.
In cancer therapeutics, this means:
Choosing treatment based on the tumor’s specific molecular alterations.
Key concept: Treatment is not one-size-fits-all.
How Precision Medicine Works in Cancer
Guided by genetic and biomarker testing
Tumors are tested for mutations or biomarkers that may determine which therapies will be effective.
Alterations can pinpoint whether targeted therapies (e.g., EGFR inhibitors, HER2 therapies, PD-1 inhibitors) should be used.
Somatic vs. Germline mutation testing
Somatic mutations
Occur in tumor cells only (not inherited).
Testing is usually done on a tumor biopsy sample.
Some assays use blood to detect circulating tumor DNA.
Germline mutations
Inherited mutations present in all cells.
Testing is done using:
Cheek swab
Saliva
Blood sample
Precision / Personalized Medicine: How it Works
Step-by-step process
Patient is diagnosed with cancer.
A tumor sample is sent for mutation/biomarker analysis.
The analysis identifies actionable or druggable targets
Examples: mutations, fusion proteins, other biomarkers.
Therapy is selected based on the actionable targets found.
This ensures treatment is tailored to the tumor’s specific molecular profile.
Mutation / Biomarker Analysis in Precision Medicine
Companion Diagnostics (CDx)
Defined as:
A medical device, often an in vitro diagnostic test, that provides information essential for the safe and effective use of a corresponding drug or biologic.CDx tests determine which patients can benefit from certain targeted therapies.
They identify mutations or alterations that are required for treatment with specific drugs (e.g., EGFR inhibitors, HER2 therapies, PARP inhibitors).
Examples of CDx tests
FoundationOne (comprehensive genomic profiling)
BRACAnalysis (BRCA1/2 mutation testing)
Precision/Personalized Medicine: Mutation/Biomarker Analysis
Key Points
Some drugs are approved only for patients who have specific genetic alterations or mutations.
These drugs often must be paired with an FDA-cleared Companion Diagnostic (CDx).
Companion Diagnostics (CDx) identify whether a patient’s tumor contains the required mutation/alteration.
This determines whether the patient is eligible for the targeted therapy.
The FDA maintains a searchable list of all cleared or approved CDx devices (in vitro and imaging tools).
Case
A 67-year-old patient has relapsed acute myeloid leukemia (AML).
The oncology fellow asks whether the patient is a candidate for enasidenib (Idhifa), a targeted therapy.
Key Question:
What information do we need to make a recommendation?
Answer
We need to know whether the patient’s AML cells have an IDH2 mutation.
Why?
Enasidenib is FDA-approved ONLY for AML patients with an IDH2 mutation, confirmed by an FDA-approved diagnostic test.
Without the mutation, the drug would not be effective and should not be used.
Precision / Personalized Medicine: Tumor/Tissue/Site-Agnostic Indications
Key Concept
Precision medicine has led to tumor-agnostic (or tissue-agnostic) drug approvals.
Tumor-agnostic indication =
The FDA approval does not specify a tumor type or location.
Instead, the drug is approved only based on the presence of a specific molecular alteration, regardless of where the cancer originated.
Important Features
Any tumor type can be treated if the required mutation or molecular feature is present.
Example: A drug may be used for colon cancer, lung cancer, or thyroid cancer as long as the tumor has the same actionable mutation.
Examples of Precision Medicine Approvals
Typical FDA-approved indications (tumor-specific)
These are approved for specific cancer types:
Tisotumab vedotin (Tivdak)
For cervical cancer that has progressed after ≥1 prior therapy.
Osimertinib (Tagrisso)
For non–small cell lung cancer (NSCLC) with
EGFR exon 19 deletion or
EGFR exon 21 L858R mutation (mutation-positive).
Tumor/Tissue/Site-Agnostic FDA-approved indications
These drugs are approved based solely on a molecular alteration, not tumor type:
Larotrectinib (Vitrakvi)
For solid tumors with an NTRK gene fusion.
Dabrafenib (Tafinlar)
For solid tumors with a BRAF V600E mutation.
DNA Packaging: Chromatin and Chromosomes
DNA → Chromatin → Chromosome
DNA alone is a double-stranded molecule.
Chromatin = DNA + structural proteins (mainly histones).
This is the default form of DNA in the nucleus.
Chromosome = the fully condensed, organized structure of chromatin formed during cell division(mitosis/meiosis).
Chromatin vs. Chromosomes
Chromatin
Definition: DNA + histone proteins that package DNA in the nucleus.
Physical State: Loosely packed (variable).
When Present: Throughout the cell cycle, especially interphase.
Function:
Allows gene expression
Allows DNA replication and repair
Types:
Euchromatin: loosely packed, transcriptionally active
Heterochromatin: tightly packed, transcriptionally silent
Chromosome
Definition: Condensed, tightly organized form of chromatin seen during cell division.
Physical State: Tightly packed and coiled.
When Present: Visible during mitosis and meiosis (especially metaphase).
Function: Ensures accurate segregation of DNA to daughter cells.
Form: Discrete structures, each containing one DNA molecule plus associated proteins.
Organization of Eukaryotic Chromosomes
Nucleosomes
Fundamental repeating units of chromatin.
Made of:
Double-stranded DNA wrapped around
Histone proteins (forming “beads on a string”).
Chromatin Structure Hierarchy
DNA wraps around histones → nucleosomes
Nucleosomes coil → chromatin fiber
Chromatin fibers loop and condense further
Highly condensed chromatin folds → chromosome
This hierarchical packaging allows:
Large amounts of DNA to fit inside the nucleus
Controlled access for transcription, replication, and repair
Nucleosome Structure
Histones
There are 5 types of histone proteins:
H2A, H2B, H3, H4 (core histones)
H1 (linker histone)
Histones contain a high percentage of positively charged amino acids (e.g., lysine, arginine).
These “+” charges attract the “–” charges on DNA’s phosphate backbone, resulting in strong DNA–histone interaction.
DNA and histones are tightly associated, enabling efficient packaging.
Nucleosome Components
Histone Core (Octamer)
Formed by:
2 copies each of H2A, H2B, H3, and H4
This octamer is the “spool” that DNA wraps around.
DNA Wrapping
~147 base pairs of DNA wrap around the histone octamer.
Histone tails extend out from the core and help maintain DNA binding.
Role of Histone H1
H1 binds DNA at the entry and exit points where DNA joins and leaves the nucleosome.
Acts like a clamp:
Locks DNA in place.
Helps stabilize higher-order chromatin structure.
Epigenetics
What is epigenetics?
The study of how cells control gene expression without changing the DNA sequence.
Epigenetic changes allow cells to regulate gene activity in a reversible, non-permanent way.
Key Features
Epigenetic modifications:
Can be maintained through cell divisions.
Can sometimes be inherited across generations.
They do not alter the underlying DNA sequence—only gene expression patterns.
Relevance to Cancer
Epigenetic changes can contribute to cancer development by:
Reducing transcription of tumor suppressor genes
→ leads to decreased protection against uncontrolled cell growth.Increasing transcription of oncogenes
→ promotes abnormal cell proliferation.
Because of this, epigenetic mechanisms are targets for anticancer therapies.
Modes of Epigenetic Modifications
Epigenetic changes regulate gene expression without altering the DNA nucleotide sequence.
These modifications are also targetable by drugs, making them important in cancer therapy.
Three major modes of epigenetic regulation:1) DNA Modification
Typically refers to DNA methylation (addition of methyl groups to cytosine bases).
Usually silences gene expression.
2) Histone Modification
Includes acetylation, methylation, phosphorylation, ubiquitination, etc.
Alters how tightly DNA is wound around histones.
Acetylation → loosens chromatin → increases transcription
Deacetylation → tightens chromatin → decreases transcription
3) Noncoding RNA
miRNAs, lncRNAs, and others regulate gene expression by:
Binding mRNA
Affecting translation
Influencing chromatin structure
DNA Modification
What is DNA methylation?
The most common type of epigenetic modification.
Involves the addition of a methyl group (CH₃) to DNA.
Methylation occurs without altering the DNA sequence itself.
Effects on Gene Expression
Hypermethylation (increased methylation):
Reduces transcription → gene is “turned off.”
Often occurs at gene promoters.
Hypomethylation can lead to inappropriate gene activation.
Relevance to Cancer
Abnormal methylation patterns can silence tumor suppressor genes.
These changes can contribute to cancer development and progression.
DNA Methyltransferases (DNMTs)
What do DNMTs do?
DNMTs add methyl groups to DNA.
Three major types:
DNMT1
DNMT3A
DNMT3B
Mechanism
All three DNMTs transfer a methyl group to the C5 position of cytosine, forming:
5-methylcytosine
This modification is key for:
Gene regulation
Chromatin structure
Stable inheritance of epigenetic marks during cell division
DNA Methylation in Tumorigenesis
How abnormal methylation contributes to cancer
Cancer is associated with two major methylation abnormalities:
1. Genome-wide hypomethylation
Leads to:
Activation of proto-oncogenes
Genomic instability
This promotes tumor initiation and progression.
2. Promoter hypermethylation
Leads to:
Silencing of tumor suppressor genes
Example: Overactivity of DNMT1 causes excessive methylation → tumor suppressor genes are turned off → cancer progression.
Example
DNMT1 is overexpressed in pancreatic cancer, contributing to hypermethylation and gene silencing.
Targeting DNA Methylation to Treat Cancer
Decitabine (5-aza-2’-deoxycytidine)
Drug class
Hypomethylating agent (HMA)
DNA methyltransferase (DNMT) inhibitor
Clinical use
Treats:
Leukemia
Myelodysplastic syndromes (MDS)
These conditions often involve hypermethylation and gene silencing.
Mechanism of Action
Decitabine is a cytosine analog.
During DNA replication:
It becomes incorporated into DNA.
DNMT1 binds to decitabine and becomes covalently trapped and inactivated.
This causes depletion of active DNMT1.
Result
Reduced DNA methylation
Increased transcription of silenced genes, promoting:
Cell differentiation
Reduced proliferation
Apoptosis
This reverses the hypermethylated, gene-silencing environment that drives cancer.
Histone Modification
Histone modifications change how tightly DNA interacts with histone proteins.
These changes regulate chromatin structure, which in turn regulates gene expression.
Modifications occur mainly on histone tails.
Types of Histone Modifications
Acetylation*
Methylation*
Phosphorylation
Ubiquitination
These modifications can either:
Disrupt histone–DNA interactions → loosen chromatin → ↑ gene expression, or
Strengthen histone–DNA interactions → tighten chromatin → ↓ gene expression
Histone Acetylation
Acetylation
Adds a negative charge to lysine residues on histone tails.
Negative histone tails repel negatively charged DNA.
Leads to:
Chromatin relaxation (euchromatin)
Increased gene expression
Deacetylation
Removes the negative charge from lysines.
DNA binds more tightly to histones.
Leads to:
Condensed chromatin (heterochromatin)
Reduced gene expression
Regulation of Acetylation/Deacetylation
HATs — Histone Acetyltransferases
Add acetyl groups.
Promote open chromatin and gene activation.
HDACs — Histone Deacetylases
Remove acetyl groups.
Promote closed chromatin and gene repression.
Cancer Connection
Imbalance in acetylation can promote cancer progression.
HDAC is overexpressed in many cancers, contributing to tumor suppressor gene silencing.
Targeting HDAC to Treat Cancer
Vorinostat (Zolinza)
Drug Class
HDAC inhibitor
Indication
Cutaneous T-cell lymphoma
Mechanism
Inhibits HDAC → prevents removal of acetyl groups
Increases histone acetylation, restoring normal gene expression
Reactivates tumor suppressor genes
Promotes:
Cell cycle arrest
Differentiation
Anti-tumor effects
Histone Methylation
Key Concepts
Histone methylation can increase OR decrease gene expression depending on:
Which amino acid on the histone is methylated
How many methyl groups are added (mono-, di-, tri-methylation)
Enzymes involved
Histone methyltransferases (HMTs)
Add methyl groups → increase methylation
Histone demethylases (HDMs)
Remove methyl groups → decrease methylation
Cancer Connection
Imbalances in methylation enzymes can disrupt normal gene regulation.
This may lead to:
Abnormal gene silencing
Oncogene activation
Cancer development
Targeting Histone Methylation to Treat Cancer
Tazemetostat (Tazverik)
Drug Class
Histone methyltransferase (HMT) inhibitor
Target
Specifically inhibits EZH2 (a key HMT)
Indication
Relapsed/refractory follicular lymphoma
Normal Function of EZH2
EZH2 adds methyl groups to histones (especially H3K27)
This increases methylation → gene silencing
Overactivity of EZH2 blocks differentiation and promotes cancer cell survival.
Mechanism of Tazemetostat
Inhibits EZH2 → decreases histone methylation
Allows previously silenced genes to be transcribed again, including:
Genes controlling the cell cycle
Genes promoting apoptosis
Effects in Cancer Cells
Restores normal gene expression
Leads to:
Cell cycle arrest
Differentiation
Apoptosis
Targeting Histone Methylation to Treat Cancer
Follicular Lymphoma (Untreated)
EZH2, part of the PRC2 complex, adds methyl groups to histone H3 at lysine 27 (H3K27me3).
This methylation leads to:
Gene repression → tumor suppressor genes (e.g., IRF4, PRMD1, CDKN1A, CDKN2A) are turned off.
As a result:
Cancer cells do not differentiate
Cancer cells continue proliferating
Bottom line:
Overactive EZH2 = too much histone methylation = silencing tumor suppressor genes → lymphoma growth.
Follicular Lymphoma Treated with Tazemetostat
Tazemetostat inhibits EZH2, preventing H3K27 methylation.
This reduces gene silencing and allows key genes to be activated, including:
IRF4
PRMD1
CDKN1A
CDKN2A
Consequences in cancer cells:
Gene activation restored
Cell cycle arrest
Apoptosis (programmed cell death)
Bottom line:
Inhibiting EZH2 reverses abnormal methylation → restores tumor suppressor gene expression → slows or kills cancer cells.