3: Amino Acids and Proteins
1/83
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
84 Terms
What is cystinuria?
A rare condition in which stones made from an amino acid called cysteine form in the kidney, ureter, or bladder.
What are the consequences of cystinuria?
Damage to the kidney and urinary system.
The linear sequence of AA is called ___________ (determines everything - the most important characteristic of a protein).
The primary structures of all diverse human proteins synthesized from ___ AA are arranged in a linear sequence determined by ___ code.
primary structure
20 AA and genetic code
Side chains/R groups _____ between AA.
vary
Sickle cell Disease
(changes what?) (how does it affect the blood?) (causes what disease?)
change of one AA, clot, necrosis
Genetic disease changes hemoglobin by a single change in one AA.
Makes sticky characteristics of protein/RBC. Clot in blood vessels like capillaries.
Sx formed: block blood flow which may cause necrosis (lack of O2)
Urinalysis for cystinuria
- what color? - had many of what cell? - contained which substance that makes crystal? - what happened to the enzyme that absorbs that substance?
reddish/brown, crystal/dimer, mutated
Urine reddish-brown (blood in urine)
Urinalysis showed many RBCs. His urine had crystals (made of hexagonal crystals of cystine - one of AA)
Cystine is a dimer that makes crystals in the urine - difficulty in excreting from the body
The patient has a mutation in an enzyme that absorbs cystine which is why this is happening.
Type I Diabetes
what happens? how to control it? what cell dysfunctional? new techniques?
high blood sugar levels, insulin shot, beta, human cadaver
destruction of pancreatic β-cells → hyperglycemia. Controlled with insulin injections (from recombinant human insulin made in E. coli and purified) or insulin pumps, though blood glucose control can be difficult.
New approaches include human cadaver islet transplantation and experimental stem-cell β-cell replacement or immunotherapies
MI description
shortness of breath, pain across chest, light-headedness. Blockage of blood flow into the heart.
General structure of AA
What happens in diff pH.
Made of alpha carbon, side chain (diff for all AA)
carboxyl group (negatively charged)
amino group (positively charged in neutral pH).
Diff pH —> changes structure:
AA changes depending on pH. pH is controlled by H+ concentration
If acidic and at a high pH = AA changes its structure
low pKa = deprotonate | high pKa = protonate.
L and D AA
Which of these forms (L and D forms) in human proteins and where is the other found
L in proteins, other in brain
L and D have similar structure but: in L-AA amino group on left and amino group is on the right
L-AA in human protein. D-AA is found in brain (neurons - not to produce protein but very specific).
Peptide bonds
What type of chemical rxn produces peptide bonds?
Between what groups of AA produce peptide bonds?
dehydration of amino + carboxyl
Water molecules removed to combined 2 AA.
Dehydration rxn (water is removed from 2 AA)
Then, AA combined to make one peptide bond.
This is Anabolic rxn
Charged AA - Basic AA (positive charged)
Arginine (R)
Lysine (K)
Histidine (H)
Acidic AA (negative charged)
D/E
Aspartate (D) (almost the same/interchangeable as aspartic acid) and Glutamate (E) (almost the same/interchangeable as glutamic acid).
Non polar Hydrophobic AA description
nonpolar, globular
Not form hydrogen bonds. Found in protein core, away from the solvent.
Globular molecules of protein - hidden in core.
Polar, uncharged AA description
Easy to be dissolved due to alcohols, thiols, or amines side chains. These side chains form hydrogen bonds and project into the aqueous phase. Outside of globular structure.
easily dissolved via hydrogen bonds
Aliphatic AA description: what is it missing, where is it located, what atoms does it have
Side chain have only carbon or hydrogen atoms
Side chain missing in side chain is oxygen. By having oxygen, easier to interact with water.
Missing oxygen its nonpolar and hydrophobic (carbon hard to interact even if H is easier).
Often located into the core of the protein.
oxygen, core, hydrogen/carbon
Cyclic AA - heterocyclic and aromatic identify which ones
what does it play a role in?
cyclic structure.
Heterocyclic: proline (P)
Aromatic AA (phenylalanine, tyrosine, tryptophan)
Cyclic AA imp role in protein structure/function, drug discovery/bioengineering.
Proline; F/Y/W; drug + protein discovery n bioengineering
Sulfur containing AA
What is each important for
Explain human insulin with peptide C importance and relation to sulfur
cysteine and methionine; start codon n disulfide; insulin produced via peptide c
Create imp for sulfur homeostasis.
Need to know these: cysteine and methionine. Methionine initiation starts codon. Cysteine is imp in developing 2D/3D structures. Two cysteines make a disulfide bridge to stabilize proteins.
Example: Human insulin is made of 2 polypeptide chains (A and B chains). Insulin is synthesized as one long stretch of polypeptide chain. Then, a long polypeptide chain makes disulfide bridges b/w cysteine. After, the polypeptide chain is cut/cleaved and releases a C peptide. Before the C peptide is gone, disulfide bridges (connect A and B chains).
Proinsulin is one chain of polypeptides that makes disulfide bridges. When the C peptide (essential) is removed, its insulin is functional. C peptide is used to determine how much the patient showing hyperglycemia symptoms produces insulin. Determines how much C peptide is released from the blood/how much insulin is produced. Sometimes, Type II diabetes releasing too much insulin so its possible to see if too much C peptide is produced or very low concentration of C peptide means they have Type I diabetes. Most imp for diagnosis is blood glucose but C peptide is to determine insulin produced.
What is oxidation and reduction? What is it in terms of cysteine?
Cysteine after making disulfide bridge --> cystine (dimer). Oxidation rxn makes cystine dimer. Reduction breaks down and makes 2 molecules of cysteine.
Oxidation (add O atoms, remove H - in this case 2) is to remove electrons and reduction (add H atoms) adds electrons.
Cysteine reduction is 2 cysteine and oxidation is cystine
Hydrophobic Interaction of Phenylalanine Causes what?
Hydrogen bonding imp for protein why?
Phenylalanine makes specific structure when 2 are stuck. (pi stacking). Hydrogen bonds are very imp to make specific structure of protein.
pi stacking; specific structure
Titration Curve of Histidine
Histidine diff pH diff structure
Histidine weakly basic (+ charge). Depending on pH, these structures are all diff. All diff structures of Histidine. High pH of 9.3 very basic and below 1.8 is acidic. pKa means acid dissociation constant.
At a pH below the pKa for each functional group on the amino acid, the functional group is protonated. At a pH above the pKa for the functional group it is deprotonated.
Adenyl cyclase structure and role
transmembrane domain with N/C/oligosaccharide - signal cascade
Adenyl cyclase imp to produce cyclic AMP. Produce 2ndary messengers for signal transduction.
This enzyme has a distinct shape. Transmembrane region have very similar structures invariant regions. C-terminus have similar structure. The other half oligosaccharide glycosylation (adds sugar) to the protein imp. Left side shows long N terminal end. C terminal end similar. The enzyme has a long N-terminal end that protrudes from the membrane. C-terminus conserved.
Imp to have variant region size function of protein/enzyme requires specific structure. Invariant Regions: These are highly conserved parts and variant regions vary.
Myoglobin vs Hemoglobin - conserved
Differences/similartieis b/w human globular protein. Myoglobin and alpha/beta hemoglobin. 2 molecules of alpha + 2 molecules of beta = tetramer of hemoglobin. Very similar sequence of AA. Myoglobin (muscle). Hemoglobin in RBC binds to oxygen. Myoglobin makes binding to O2 in muscle (cardiac/skeletal). Called conserved AA (primary structure).
for o2 binding
Why do all human globin proteins show similar primary structure and what does this mean to their functions
even if valine and isoleucine diff, similar characteristics. Myoglobin binds to oxygen and hemoglobin binds to o2 well --- for o2 binding, sequence is imp in primary structure.
for o2 binding
Electrophoretic separation of CK
CK2 (released to blood from heart muscle) higher peak in cardiac when damaged vs CK3 in skeletal
Blood tests and the case of myocardial infarction. Chest pain - blood testing done to check specific protein - creatine kinase. Creatine Kinase is an enzyme to add phosphate group to specific protein. Its imp to regulate o2 in muscle, brain, and other tissues by using ATP. Without having heart attack, person shows 2 isoenzymes of creatine kinase.
Big peak of CK-3 (skeletal muscle) and CK-2 is basically found in cardiac muscle. Creatine kinase released from ur heart to blood during damage and normally stay inside of cardiac muscle (myocardium). If someone epxeriencing MI, heart muscle is damaged. normal condition - specifically found in myocardium of heart so heart muscle damaged since should stay inside not released in blood.
High peak: CK-3 in skeletal and cardiac but CK-2 is only in cardiac so when its damaged, creatine kinase released in blood and heart muscle damaged.
Creatine kinase isoenzymes role and how can they be apart
adds phosphate to creatine (controls ATP/oxygen level in brain, heart, tissues). What is isoenzymes? peaks of creatine kinase but a lil diff primary sequence of AA. They do the same chemical rxn. Distinguished by instrument by HPLC or system even when they do same thing (add phosphate to creatine). Have a lil diff molecular weight and enzyme.
same chemical rxn; by HPLC
Primary structure of human insulin
proinsulin synthesized in one chain. Disulfide bridges. C-peptide needs to be attached but after the structure is made, the C-peptide is cleaved but useful to see protein insulation by counting C-peptides in blood. Diagnosis of diabetes is done by blood glucose levels.
disulfide with C-peptide
Post-translational modifications of AA
After proteins are synthesized, modifications occur. The carbohydrate (sugar) added is glycosylation. Imp - glycation is adding sugar to protein. Normal function of protein needs sugar - glycosylation AA.
Lipid added. AA modified by oxidation and carboxylation.
glycosylation, lipidation, carboxylation, oxidation
Sickle cell crisis - how are tissues impacted and is sickle cell hereditary (what happens in terms of genetics?)
Sickle cell crisis - low O2 levels. Tissues dead by losing blood flow. Genetic condition where gene mutated.
Cystinuria - treatment
Cystinuria - CRISPR hopefully to change mutation sequence by taking cells from patient and back to patients body to change- nanoparticle delivered
MI - how to test it (2 ways)
Case about MI. Diff types of tests used like creatine kinase. Small peark becomes bigger as creatine kinase expressed in myocardium. Peak appears means heart muscle damaged. Troponin to see if patient experiencing MI.
creatine kinase and troponin
Type 1 vs Type 2 diabetes
Glycation Hemoglobin Effects
Type 1 is autoimmune and Type 2 resistant to insulin
glycation (negative effect). hemoglobin shouldn't bind to sugar but it happens if blood glucose levels are high.
The _____ structure of a protein determines the way a protein folds into a unique 3D structure called native ______
primary, conformation
The tertiary structure consists of structural ______. What is it made of and what can it bind to?
motifs. consist of domain (specific sequence that makes binding to ligand)
Specific molecules called a ____ may bind to certain structural domains
ligand
Protein denaturation is the loss of _____, _____, and _____ structure. Occurs after ___ synthesized with no change of ______.
quartnerary, tertiary, and secondary by changing temp/pH/concentration of ion/etc. Denaturation occurs after protein synthesized with NO change of AA.
Tertiary vs quaternary - how many polypeptide chains?
quaternary - multiple polypeptide chain
Tertiary - one polypeptide chain
Amyloidosis
conformational change to protein?
what builds up?
what disease?
Beta amyloid of alzehimer disease - condition of normal protein - cut/truncuated. Amyloid accumulated in brain is alzheimer. Amyloid protein that makes abnormal accumulation in diff tissues. Many organs r damaged. Called Amyloidosis bc of abnormal protein/accumulation of diff organs.
accumulation of amyloid - alzheimer
Hemoglobin (how to check glucose levels)
tested in blood test. Easy to check and quick. % shows how much glucose a person has. Hemoglobin can be glycated (sugar is added to specific AA of hemoglobin). This shows how much glucose in the blood since protein is glycated. Sugar is automatically added without intention. Person with a high % means person has high level of glucose. If low %, person doesn't have lot of glucose in blood. To determine if person has hyperglycemia.
glycation
Name of bonding b/w AA in polypeptide chain?
peptide bond. Dehydration makes it.
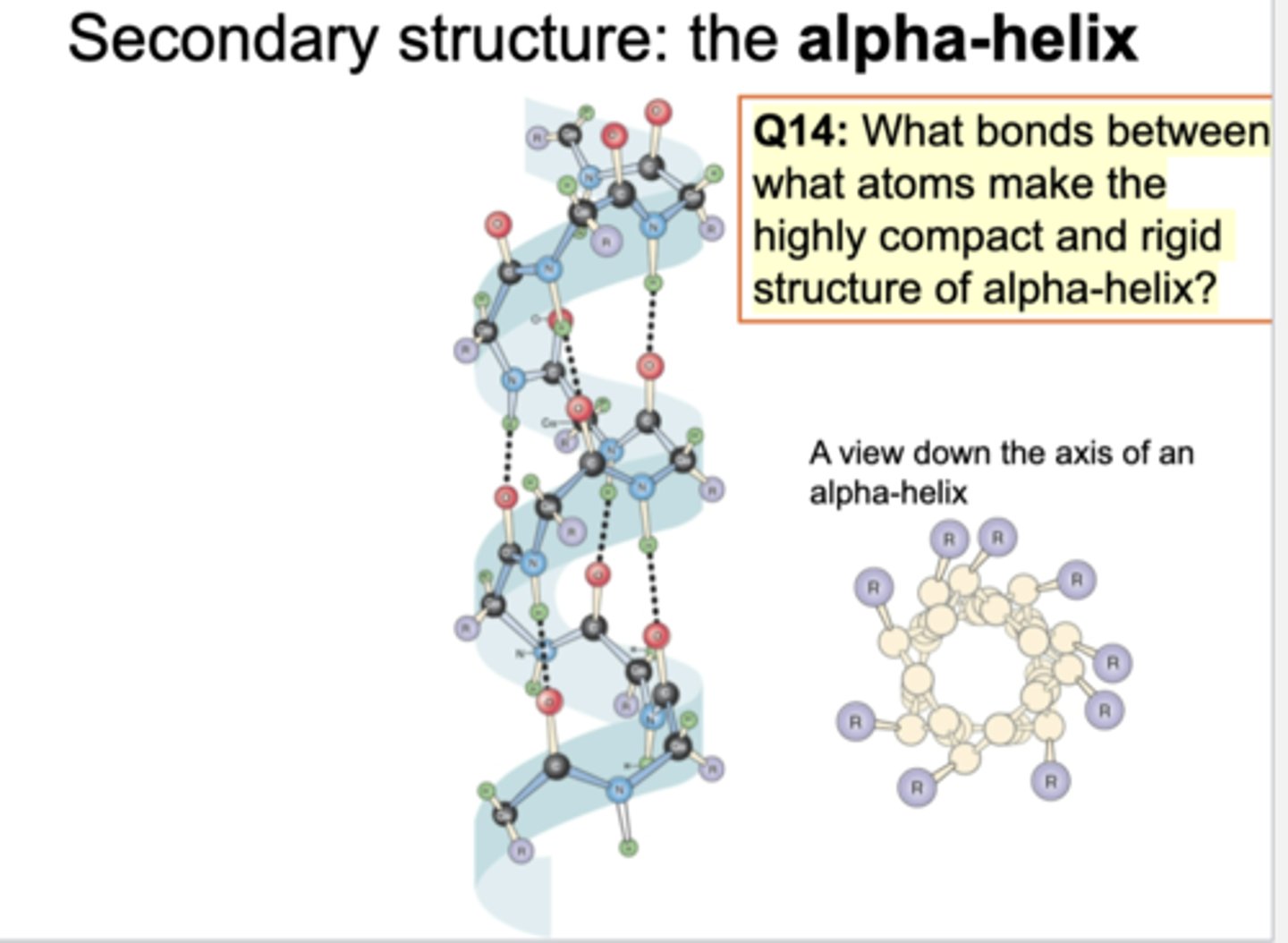
2nday structure of alpha helix
what are outside?
what bonds?
To make this structure, spiral is seen. Hydrogen and oxygen making some association/connection (hydrogen bonds). To make alpha helix, it is essential to make multiple hydrogen bonds. By looking at top, see spiral alpha helix structure. Side chains r outside of backbone structure.
hydrophobic R chains; hydrogen
2ndary structure of beta pleated sheet
parallel and antiparallel - which one more stable?
Beta pleated sheet. Two types both are beta pleated. One is antiparallel and other is parallel. Antiparallel (see strings and each string is in opp direction and is called antiparallel). Parallel means each string is same direction. Parallel means slightly bent. Parallel beta sheet are less stable (unstable) and antiparallel are more stable. Oxygen and hydrogen making specific bond. Straight nice way to make hydrogen bond in antiparallel but in parallel, they are bent not straight/aligned.
antiparallel (straight)
Beta turn
what bonds make the hairpin looop b/w 4 AA residues?
Repetitive or not?
small structure imp to make 2ndary structure. Small turn making antiparallel direction. Hydrogen bond imp to make beta turn. Non-repetitive.
Tertiary
combination of diff type of 2ndary structure. One polypeptide chain. Multiple alpha helices and beta sheets (parallel or antiparallel) seen (unstable - bent or stable - not bent). Some turns involved b/w them.
comb of alpha, beta sheets, turns, folds
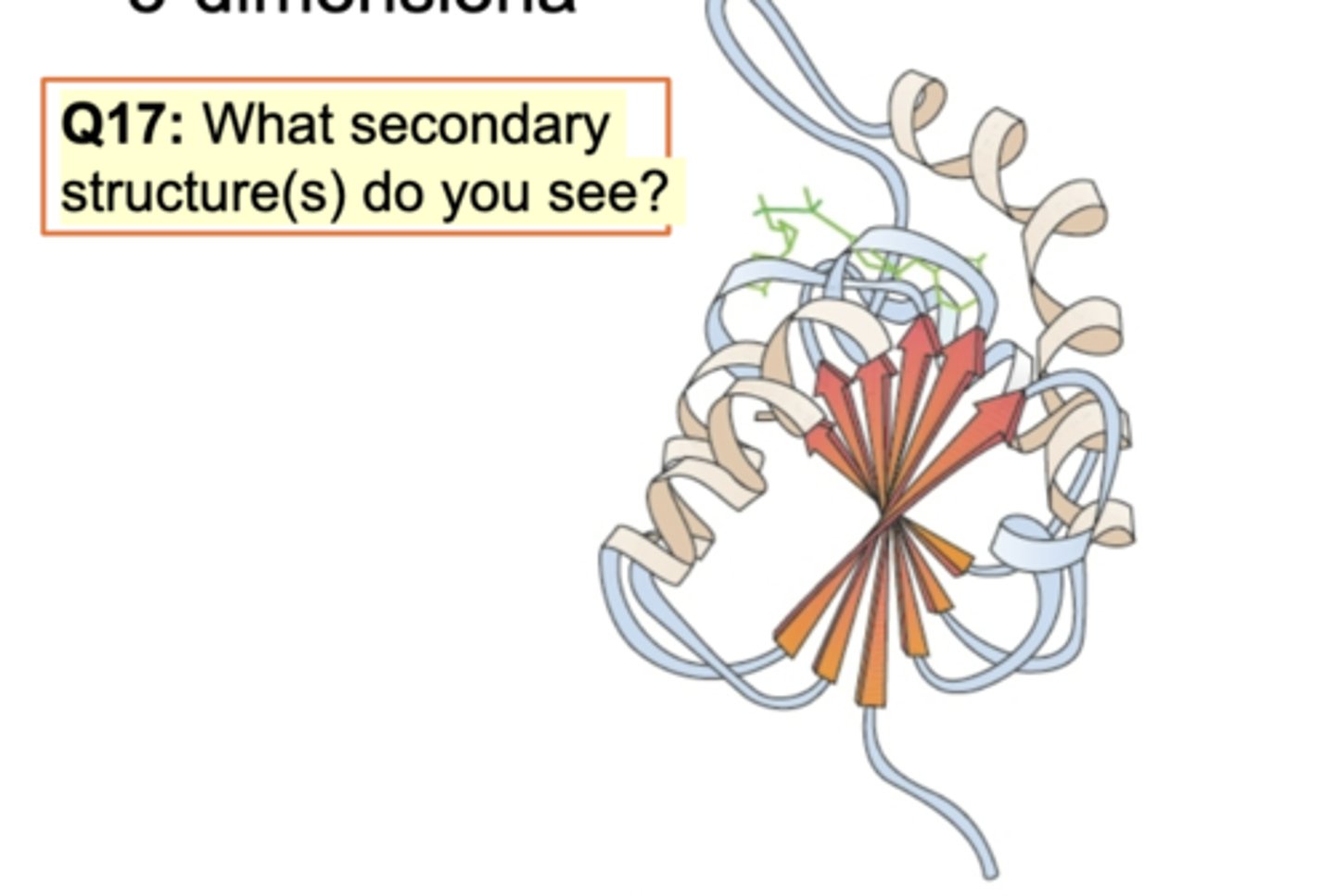
Folds in Globular Protein (G-Actin)
Diff domains in one protein G-actin. Diff secondary structure and G-actin binding to ATP is required. Alpha helices and antiparallel beta sheets and beta turns are connected.
Domain: G-actin bind to ATP
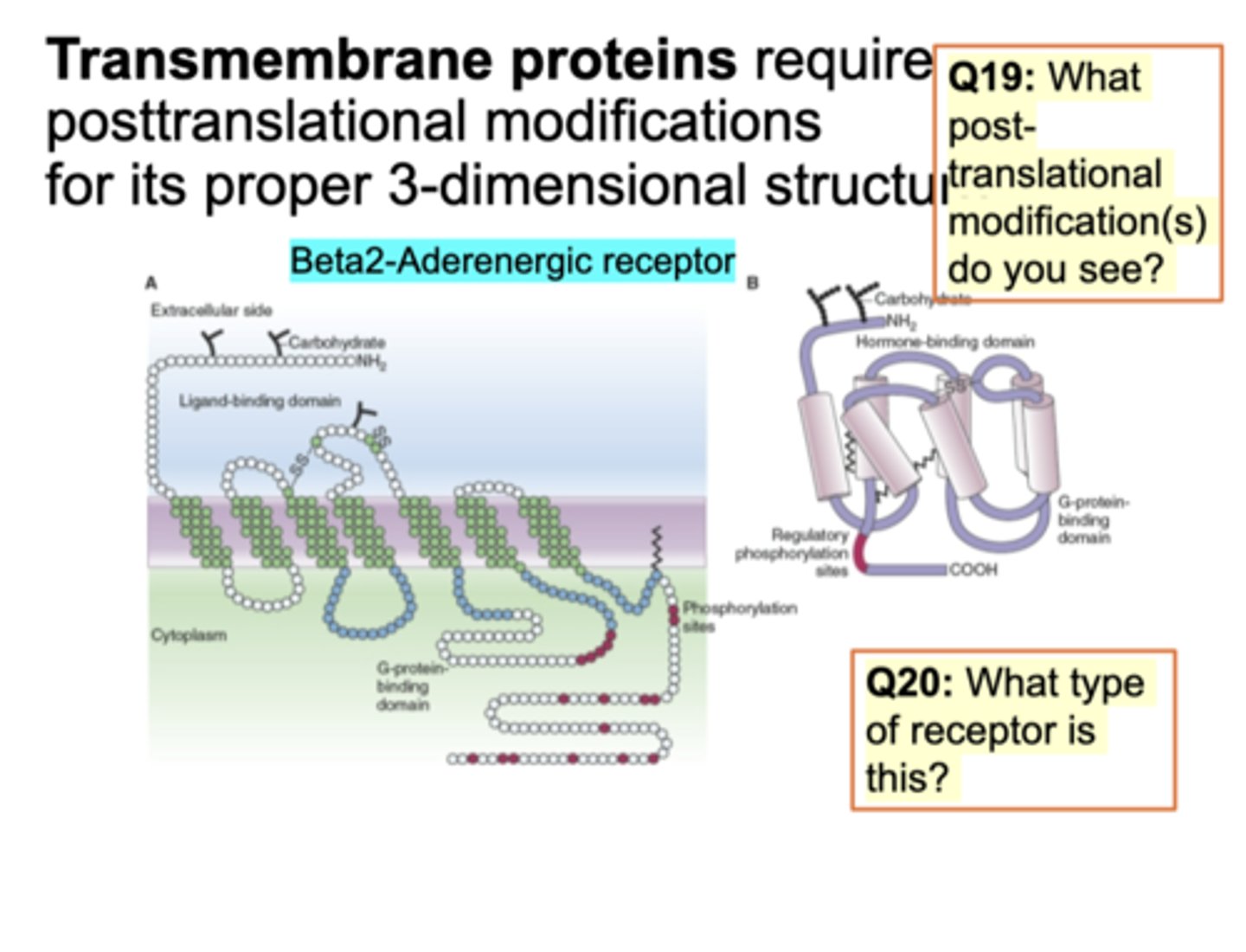
Transmembrane Protein: terminus and GPCR
protein is spanned multiple times. Protein is hydrophobic to make this structure and embedded. Diff types of C-terminus and N-terminus. Adding sugar/phosphorylation at C-terminal end. G-protein binding domain - small protein that binds to GDP and hydrolysis (exchange b/w GDP and GTP). Receptor is B-adrenergic receptor to make epinephrine/norepeinpehrine. Structure imp.
N/C; GDP for GTP and B-adrenergic for epinephrine/norepinephrine
Question on slide: What post-translational modification seen? glycosylation seen (carb addition), phosphorylation, and sulfur (disulfide bridge). What type of receptor is this? Tyrosine kinase/ion channel recceptor - GPCR. This receptor binds to G-protein.
What type of receptor is this? Tyrosine kinase/ion channel recceptor - GPCR. This receptor binds to G-protein.
Quartnerary structure, hemoglobin
multiple polypeptide chain assembled. Many proteins function as dimers, tetramers, or oligomers. Each protein has subunits. Subunits of a particular protein always combine in the same number and in the same way. Because of this, one AA chain can affect a lot like sickle cell disease and quartnerary structure can be destroyed.
one AA can destroy quartnerary
Quaternary Terms
Homo
Hetero
Protomer
Oligomer
Multimer
homo - protein made of identical subunits
Hetero - protein made of non-identical/diff subunits
Protomer - structure composed of non-identical subunits
Oligomer: multi-subunit protein composed of identical subunits
Multimer - many subunits of more than one type
Quantitative Kinetics (Ka vs Kd)
Oxygen must be released at one point: Kd and Ka=1/Kd
Ligand and domain makes binding. How oxygen makes diff types of affinity to protein like hemoglobin/myoglobin. Understand kinetics of protein binding - association constant Ka. Hemoglogbin binding to oxygen - have oxygen binding site to hemoglobin. Oxygen is a ligand. Constant for binding reaction b/w ligand and protein is Ka.
Rxn maintained. Most chemical rxn are reversible from left to right and right to left. Oxygen and hemoglobin binding/diassociating. If oxygen binds to hemoglobin forever, u can't use oxygen so oxygen must be released (reverse rxn). Rate of rxn is Ka (association constant) and Kd (dissociation constant). Ka = 1/Kd. If you see big Ka like hemoglobin bind to O2 strongly if u see low Ka, hemoglobin isn't binding to O2 like in sickle cell disease. Low kd.
2 diff types of ligands A and B bind to same receptor. Hemoglobin binds to o2, co2, nitrogen monoxide (dangerous). multiple ligands can bind to one thing. Kd for A is 10^-7 M and for ligand B is 10^-9. Which ligand has a higher affinity for the receptor?
Ligand B cuz its smaller so larger Ka.
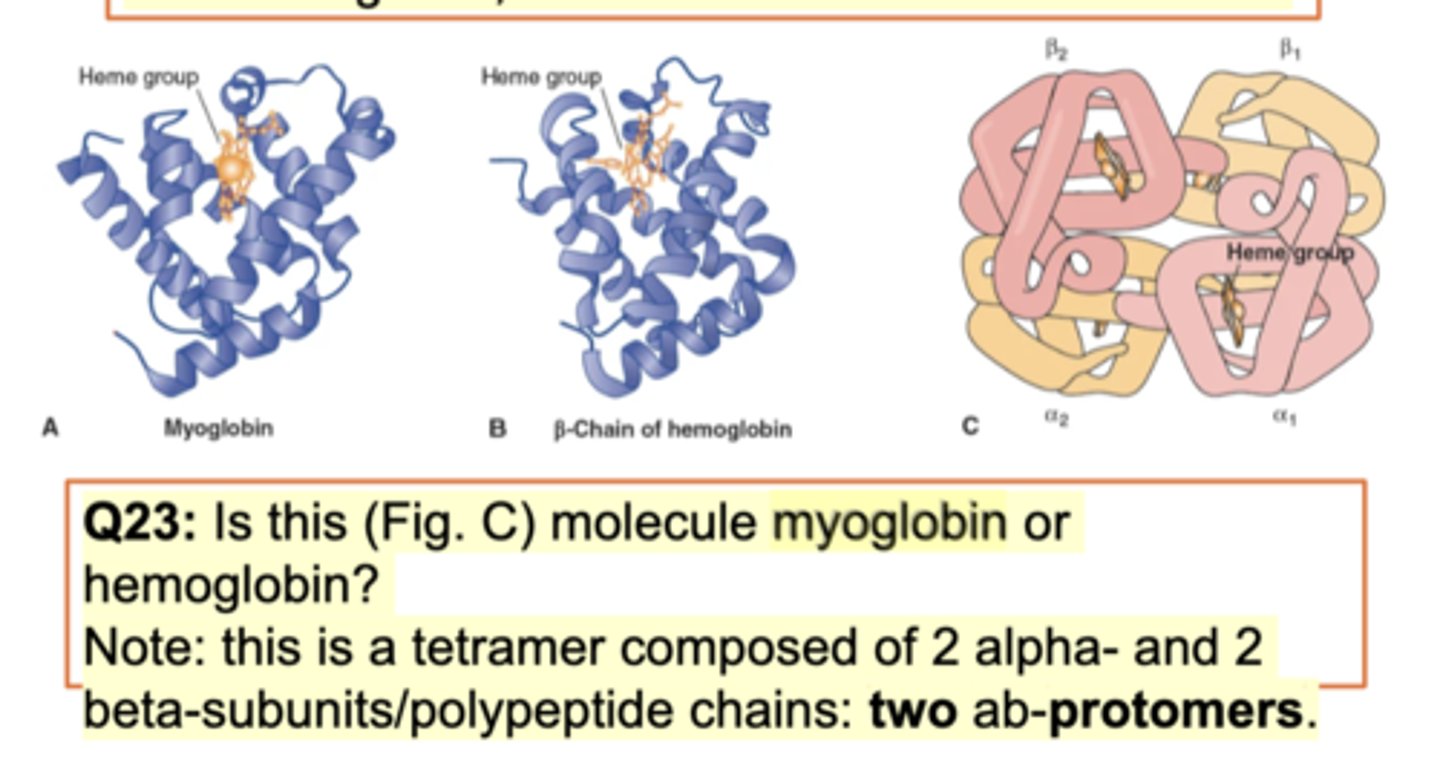
Structures of Myoglobin in Comparison to Hemoglobin
one globulin vs 4 globulin (2 alpha and 2 beta) - both have prosthetic group (bind to iron)
myoglobin muscles. B-chain of hemoglobin pic seen (tertiary structure -very similar to myoglobin). Diff is function b/w myoglobin and hemoglobin. Both bind to o2 but different.
Structure diff - Myoglobin works as a monomer but hemoglobin binds to 3 diff subunits (2 alpha and 2 beta sunits makes 1 tetramer of hemoglobin). Hemoglobin is quartnerary. Myoglobin - monomer. Hemoglobin - tetramer. Works in diff tissue/cell and a lil diff shape. Both have heme group (prosthetic group - chemical structure that binds to iron to make the hemoglobin/myoglobin functional).
Oxygen binding
Each heme can bind to an oxygen ____.
2 oxygen (o2).
Tertiary structure: 8-alpha helices aka globin fold: no beta sheets
Helices make hydrophobic O2 binding pocket containing tightly bound heme with an iron atom in center.
Organic ligand bound to protein - prosthetic group (heme). Doesn't disassociate - strongly bound. Heme isn't a protein but an ORGANIC ligand binding to hemoglobin/myoglobin.
O2
Apoprotein vs holoprotein
Explain heme group - what happens if degraded?
Apo - prosthetic group not attached and Holo - attached
diassociates
Depending on prosthetic group, before making complex structure, it isn't bound to heme. Once bound to heme/hemoglobin/myoglobin, it doesn't diassociate. If still not bound/middle of assembly, without prostethic group is called apoprotein. After binding to prosthetic gorup, its called a holoprotein (complete set). Once bound doesn't diassociate - very imp.
Tightly bound prosthetic group (planar porphyrin ring of heme) attached to fe2+ doesn't disassociate until protein degraded.
Myoglobin vs Hemoglobin
Type of curve
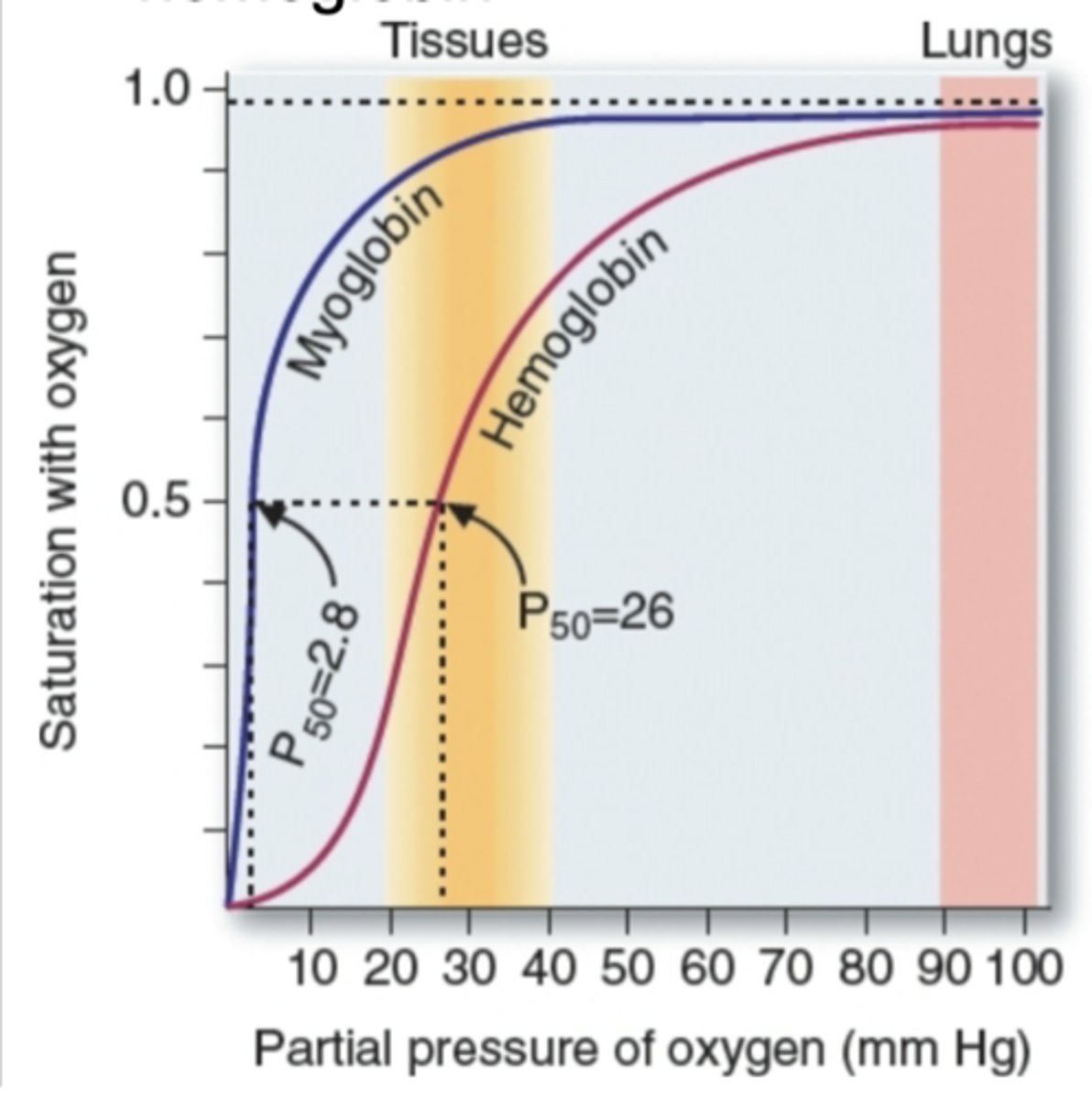
difference between P50 for oxygen-binding characteristic
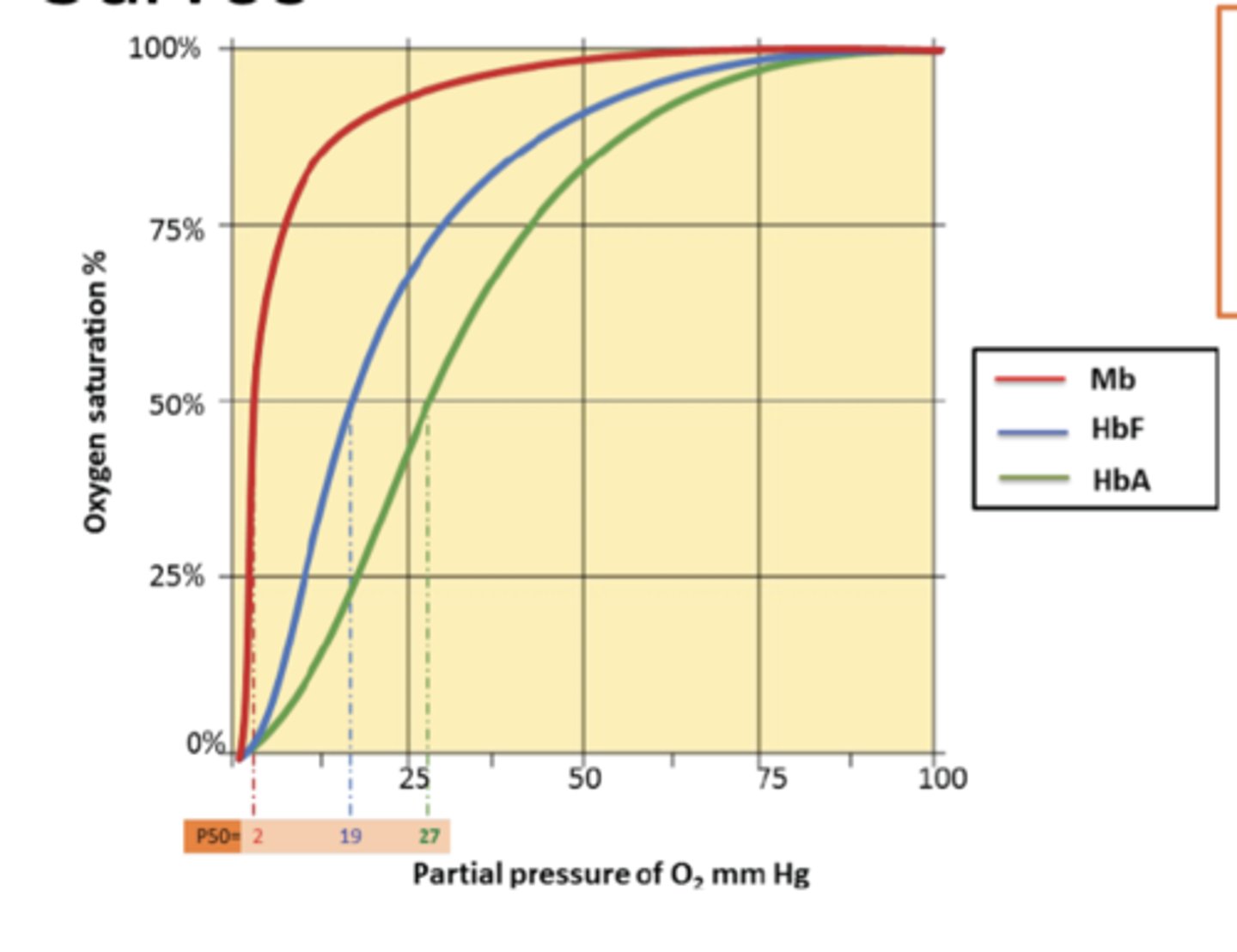
hyperbolic and sigmoidal; lower po2 for myoglobin
Myoglobin/hemoglobin diff abt function. Partial pressure of O2 means atmopsheric air contains carbon dioxide, oxygen (21%), nitrogen. Diff types of gas have atmospheric air. Oxygen is called partial pressure (mmHg). By increasing more o2 concentration is hemoglobin/myoglobin makes binding of protein to o2 and y-axis is saturation with o2. How many molecules of myglobin is saturated (binding to o2). What type of curve is myoglobin graph? hyperbolic curve (very low %/pp of O2 makes a huge binding efficiency of oxygen - myoglobin binds strongly/quickly). Hemoglobin is slower - more partial pressure of O2 needed to make saturation - sigmoidal curve. Makes a diff in binding diff is the curve. P50 means 50% of mygolobin saturation and 50% of hemoglobin saturated. To compare kinetics, 50% of each molecule bound to o2 used. By looking at x-axis, pressure diff: very low pressure can make myoglobin saturated quickly. (see graph in detail)
Sickle cell: long fibers, AA replacement, 2 types, knob
polymerization; valine; stick
Changed in AA makes diff. SIckle cell diseaee patient - polymerization of hemoglobin - changes shape of RBC. Each RBC contains hemoglobin molecules. By changing shape of hemoglobin molecule in sickel cell makes structure. Happens when RBC and hemoglobin not binding enough to O2 (deoxygenation - abnormal shape). If sickle cell disease binding to O2, shape normal. One AA change makes a huge diff bc polymerization occurs. Mutation makes valine AA exchange - makes polymerization of hemoglobin in sickle cell patients. U can see normal shape and crescent shape in blood smears. Why 2 types? Not all RBC crescent shape as some normal bc all these RBC have same sequence of AA. Only 50% showing crescent shape. Other normal even tho RBC have DNA sequence.
In sickle cell disease, the substitution of glutamic acid with valine in the β-globin chain primary structure of hemoglobin creates a "knob" or hydrophobic patch on the surface of deoxygenated hemoglobin. This patch allows the abnormal hemoglobin molecules to stick together and form long, rigid fibers, causing red blood cells to sickle and become rigid. This sickling reduces the flexibility of red blood cells and can lead to blockages in blood vessels.
Polymerization and Crescent Relation to Being Stuck
Only when RBC not binding to O2, polymerization of RBC change. See complex and crescent shape don't work - RBC gets stuck in capillaries and no O2 binding.
stuck in capillaries
H+, 2,3-BPG, CO2 binding affect affinity by lowering or highering?
Diff things binding to hemoglobin like hydrogen ions and 2-3 BPG (naturally produced in human body n bind to hemoglobin) + covalent binding of Co2. - lower
lower
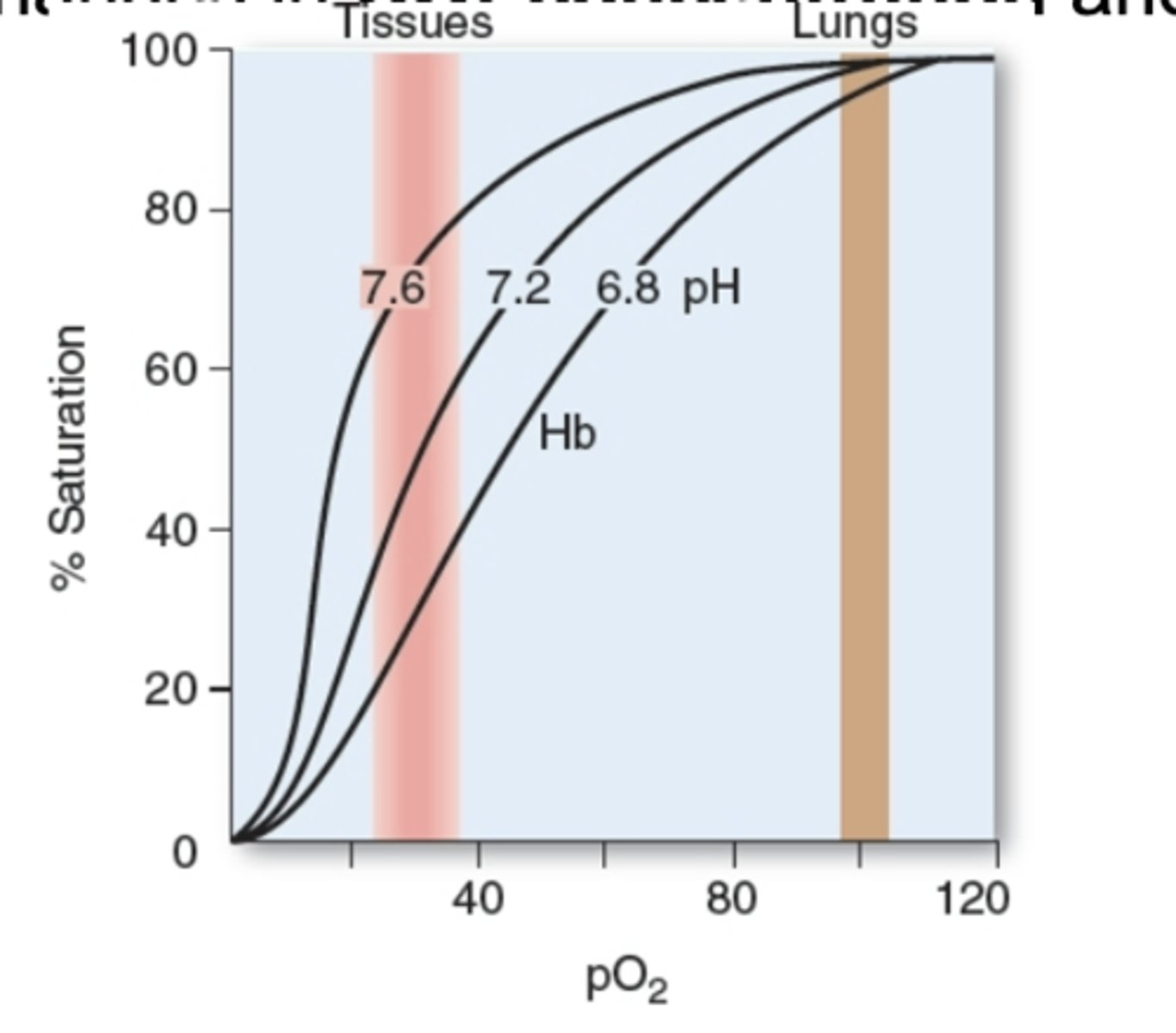
Bohr effect - pH acidic
Hemoglobin o2 binding is affected inversely to acidity and concentration of Co2. When CO2 goes higher, oxygen binding is lower. Acidity increases so pH lower. Binding to oxygen of hemoglobin will go lower. Shift in curve. Depending on pH see shift/curve. Same Hb by changing pH and going acidity higher, then affinity of oxygen goes lower. By lowering pH, increasing acidity. See curve.
right shift
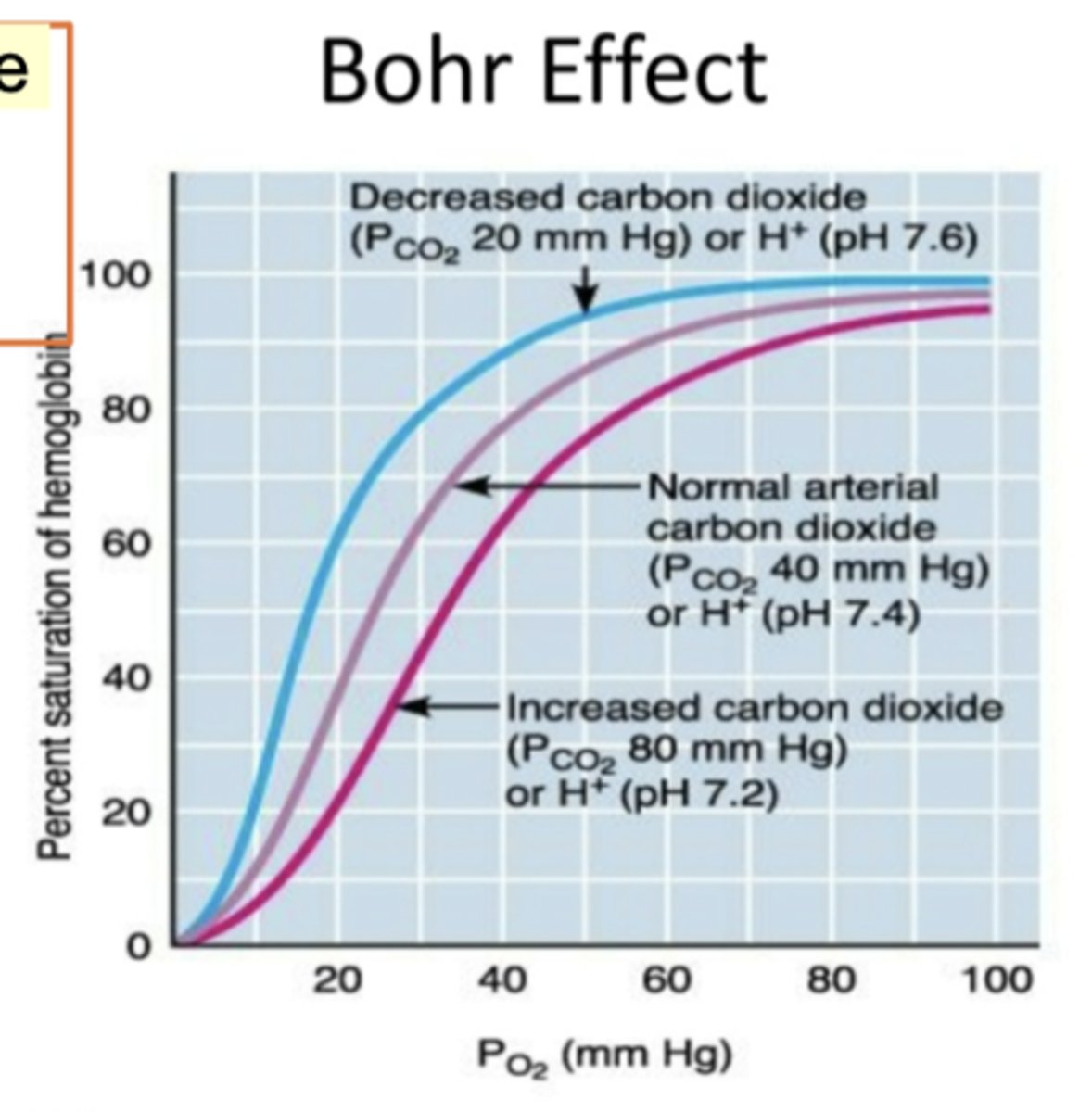
Bohr Effect - Co2 relation
Acidity increased and then pH goes lower. When pH goes lower, acidic. Affinity of hemoglobin to oxygen goes lower. High pH is alkaline - basic condition - better for binding to oxygen. Graph shows partial pressure of O2 (mmHg). Y-axis shows saturated hemoglobin. Pink line is showing increased carbon dioxide and oxygen binding of hemoglobin. The blue line is showing decreased carbon dioxide so what is shown in the graph - oxygen partial pressure increasing from left to right. Pressure of Co2- higher level of Co2 in pink and decreased PCo2 is blue. low cocnenrtration of Co2 - makes better binding of hemoglobin to oxygen. Lower the cocnentration Co2, the higher the affinity for O2.
left shift
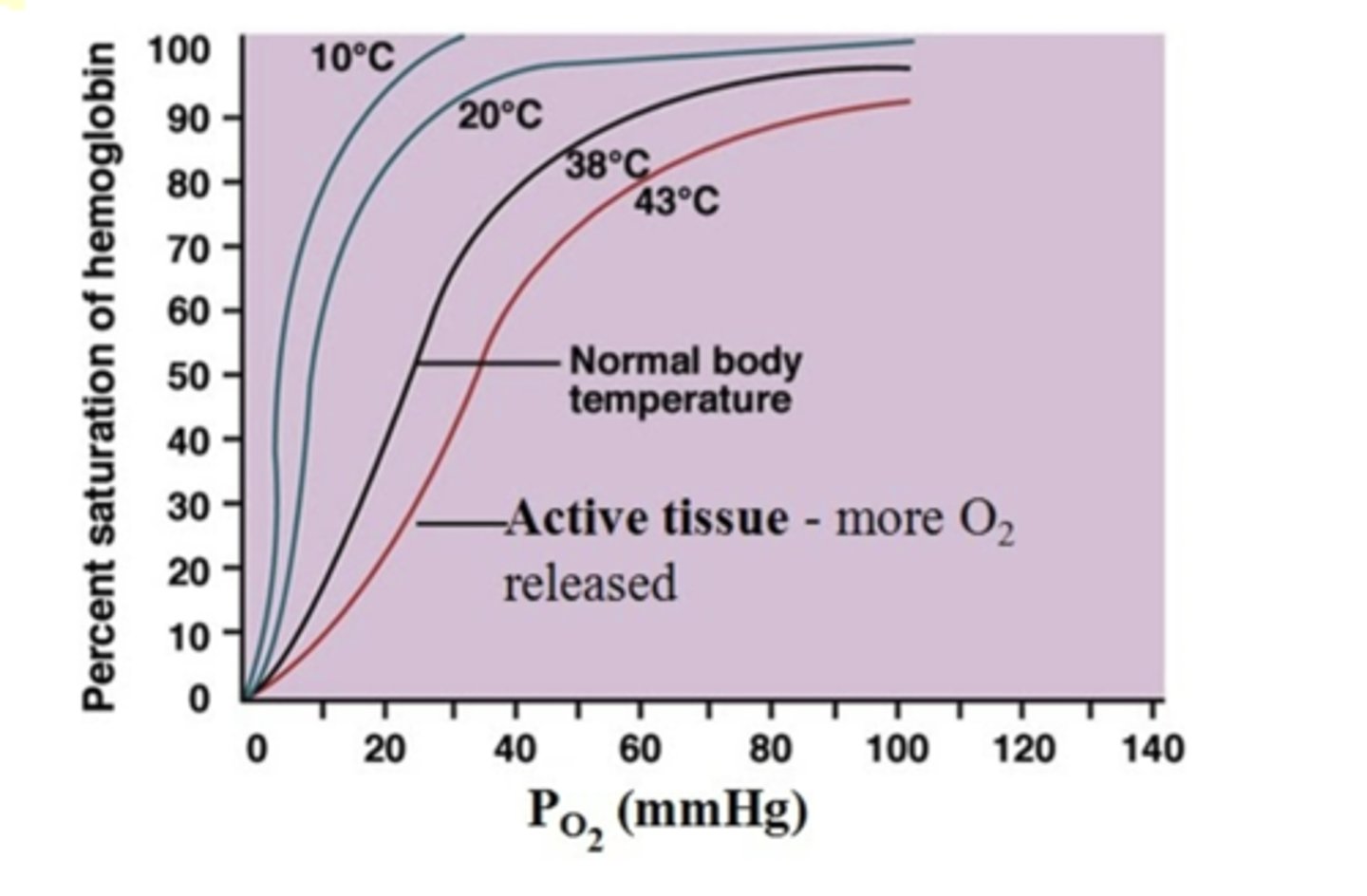
Temp
PO2 on x-axis and % saturation of hemoglobin on y-axis. See diff - the higher temp - pink line. The higher the temp, lower the affinity for O2.
lower affinity
Mygolobin -red, fetal hemoglobin - blue, adult hemoglobin - green explain saturation curve graph
partial pressure of o2, oxygen saturation. Myoglobin best binding to o2, then fetus is better. Fetus needs to grow faster so more o2 compared to adults. Fetal hemoglobin diff development/size but same DNA is same used.
myoglobin, fetus, adult left to right
R vs T hemoglobin
High affinity of R state to low affinity T state of hemogogin. Deoxy and oxy can be seen in pic. Protein structure dynamic.
Immunoglobulin structure and importance as defense
What is ligand?
Immunoglobulin are imp to protect humans from infections. 5 diff classes of immunoglobulin depending on what ppl are fighting against. Each antibody has common structure (2 identical small polypeptide Light chains) and two indentical large polyepptide chains (heavy Hchains). 2 heavy + 2 light= 1 immunoglobulin structure
Ligand: antigen
for antigens (ligand)
Variable vs constant region function
2 light chains in yellow and blue chains (heavy) makes a Y-shape. Disulfide bridge makes Y-shape. Variable (V) and constant regions (C) of immunoglobulins. V makes changes of sequence which is why diff antibodies against infection. Body can produce diff sequence/shape by using variable region. Constant regions are shape sequence. Depending on what pathogens, diff # of antibodies bc of all variable chain of light/heavy chain. How so many diff comb of variable region?Recomb by switching sequences.
variable changes (for infection and binding) while constant shape sequences
Heat Shock and nascent polypeptide - using what mechanism via what rxn
Hsp70 and Hsp60 imp for protein folding. All proteins need help by heat shock proteins. When individual exposed to higher temp, environment changes or some emergency - specific protein produced. Chaperone are proteins that help make up each protein shape. Correct conformation of each protein made by Hsp60/70 binding to nascent polypeptide chain (newly synthesized) and correctly folded proteins made by ATP hydrolysis.
correctly folds by ATP hydrolysis
Fibrous proteins - structure and what molecules present
Collagen is made of protein. Glycines present n close contact to make specific shape/translational modifications imp for shape.
Hydroxylation of proline/lysine in collagen via what n through what bond
Hydroxyproline (hydrogen bond) used to stabalize and cross linking b/w collagen molecules can further stabalize collagen fibril.
Denaturation - through what means
Changes of structure/function and denaturation by means like nonenzymatic modification, temp, pH, solvent, prions ( by misfolding not virus or bacteria but infectious protein), protein misfolding.
Modification: glycation vs glycosylation
Glycation naturally vs glycosylation is enzymatic
Modificcation of specific proteins. Glycation is nonezymatic addition of sugar. Glycolsation is an enzymatic process where sugars are covalently attached to proteins or lipids, modifying their function and stability. Glucose is present at high concentration but its a kind of accident if glucose is there without enzyme. 2 specific common enzymes works since protein needs glycosylation - quickly n efficienly so enzymes must work. Glycation naturally happens - protein oxidized or glycated even if its a bad thing. Withotu enzyme, how many sugar is added depnds on glucose cocnentration. Higher glucose concentration around protein - higher glcycation. With enzyme, enzyme increases addition of sugar. More and more sugar but no enzyme required - glucose naturally added by chance. More glucose, more glycation.
Physicians use HbA1c - why?
Why does it reflect blood glucose level only for past 4 months?
glycated - random. RBC last for 4 months.
Physicians use HbA1c - specific AA glycated if glucose is present. If your blood glucose controlled, then not many chances to be glycated. High glucose in blood can make more glycation so simple blood test can determine how much glucose present in the past. More glycation can occur.
Why is HbA1c used to determine blood glucose level? very low concentration of glucose can glycate. Without enzyme nothing happens so just by chance - proportional constant - good way to present how much glucose. simply depends on concentration - without enzyme - many glycation. Enzyme makes efficient glycation is bad but no enzyme is good measurement.
Why does it reflect blood glucose level only for past 4 months? Hb1Ac check glycation - 4 months ago. Since RBCs last for about 4 months, HbA1c reflects glucose levels over this period. The test shows the average glucose levels over the lifespan of the RBCs.
Hyperglycemia - glycated
AGEs - impact old
AGE aggregates (glycated + collagen cross links) so control glucose levels
Hyperglycemia have high glycated. Person has low insulin, glucose level high so much higher glycated to hemoglobin.
Better to control glucose level due to AGEs. Glycated proteins and collagen modified by nonezymatic oxidation and form additional cross links lead to AGE aggregates which accumulate with age eveein in ppl with normal glucose levels
Bond disruption in proteins by pH, ___, and _____
at diff pH levels (ionic n hydrogen disruption), solvent, temp
Many effect protein function like pH, temp, solvent that disrupts ionic/hydrogen/hydrophobic bonds. At low pH, ionic bond and hydrogen forms and would be disrupted. Alkalaine - hydrogen and ionic disrupted. . Diff pH = diff disruption
Stomach acid and albumin change conformations
Stomach acid - native conformation of ingested protein better access from digestive enzymes
albumin changes in egg.
SDS - polypeptides
Examples: hydrophobic molecule capable of denaturing & protein precipitates dissolved by amphipathic agents
breaks to straight line; long chain FA; urea/HCl/SDS form hydrogen + hydrophobic interaction
SDS used to do separate proteins. Binds to polypeptide chain and denaturation to make straight line structure - disrupts globular structure. Larger polypeptide binds to more molecule - slower migration.
proteins precipitates dissolved by amphipathic agents like urea, HCl, or SDS that form extensive hydrogen + hydrophobic interaction in protein
Hydrophobic molecules denature proteins by disturbing hydrophobic interaction in protein. Long chain FA can inhibit many enzyme-catalyzed rxn by binding nonspecifically to hydrophobic proteins and disrupting hydrophobic interactions.
Prion affects which part of body? acquired how? infectious or not?
brain; from infection (mad-cow) or sporadic or inherited like Creutzfeldt-Jakob; infectious but not as bacteria/virus
Prion is a protein and it makes a mess and effects protein around - bad protein. If prion doesn't cause protein misfolding, its normal. Protein folding is dynamic. Proteins always making movement and sometimes makes misfolding. Somethig goes wrong. Could happen to prion. One makes misfolding interact with enighboring protein. Normal prion affected by misfolded protein so likely infectious. Most don't have misfolded proteins but if something wrong happens, some may have disease if prion disease and ate beef- beef prion affected human prion (Jakob disease) Not infectious agent. Protein infecting - prion. Prion disease makes misfolding - aggregation. Highly expressed in gray neurons. Misfolded prion? One of them is Jakob disease - one affected by Mad Cow Disease (beef). Mutations - familial Jakob.
-neurodegenerative (can misfold others)
-prion acquired from infection (mad cow disease) or sporadic or inherited mutation like Creutzfeldt-Jackob disease
Aggregation causes ____
cadaver use
Normal vs abnormal forms in abbreviation - conformation change or same?
Gene mutation can result in prion disease as well. Eating cow - having prion disease and individual with prion disease. If person used to extract insulin and cadaver person affected by prion could be translated. Diff causes by basics is prion misfolding. Normally found in brain... function unknown. Causes misfolding. # of misfolded prion is smal which is ok. More and more misfolding makes aggregation of protein. Aggregation msifolded prion can't be degradaded due to precipiation .. neurons die due to insoluble precipates.
infectious transmission (prion) via cadaver for insulin transfer
PrpC - normal and disease cauisng form is PrPSc - disease scrapies reported - same AA
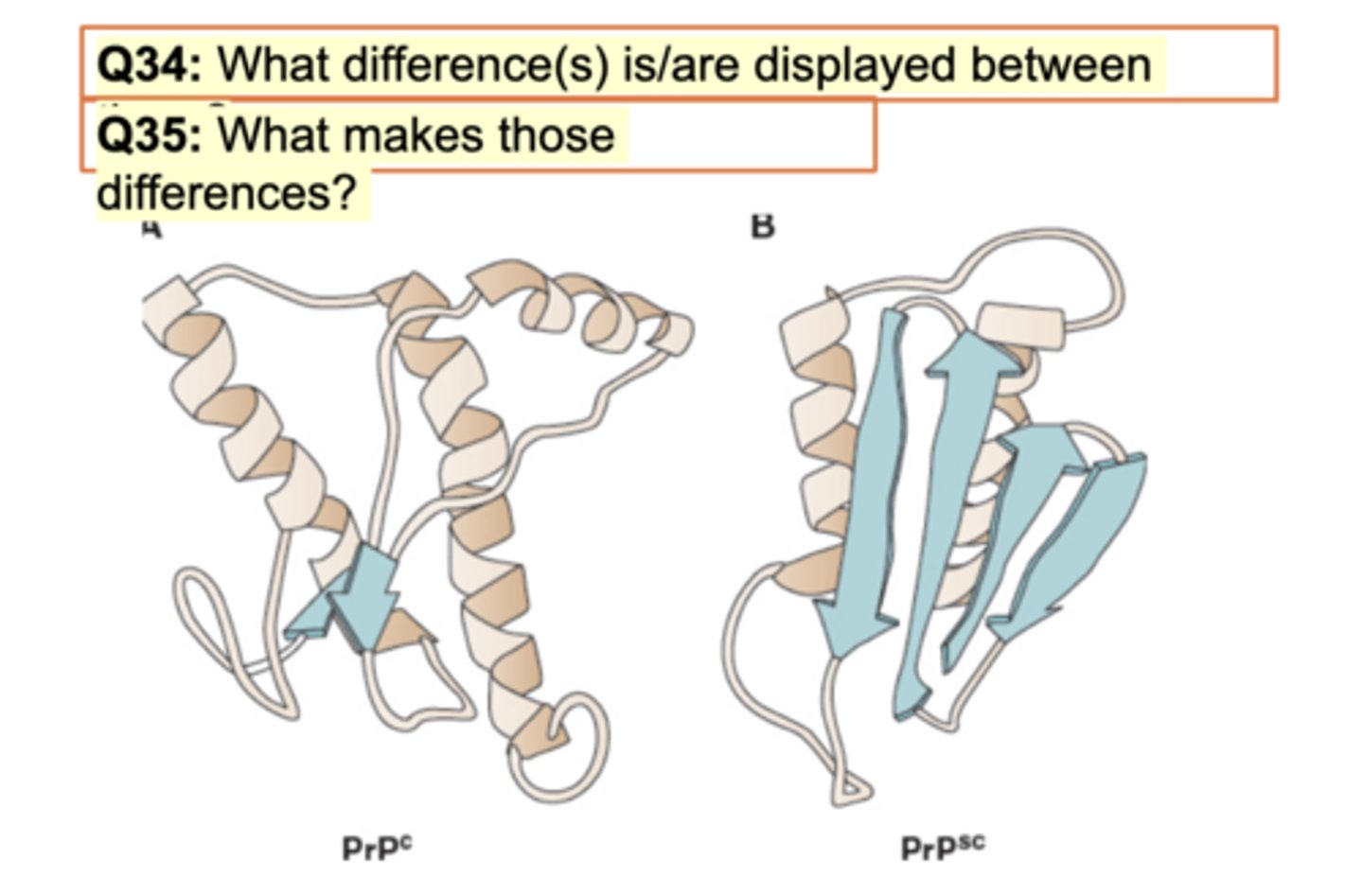
Diff in image for prion on slide
Normal prion PrpC vs abnromal on right. What difference? Same sequence but lil difference. Pink showing alpha and blue showing beta sheets. Right is opp direction antiparallel. BEtween both is big diff in beta sheets. Right is increased difficulty to dissolve so precipiatation/aggregation. SOme ppl have genetic disease but if don't have them, misfolding seen here occurs.
What makes those differences? Misfolding.
right has more antiparallel sheets: misfolding
Energy for prion misfolding
To make misfolding, more energy is required. More energy doesn't make sense so usually doesn't occur but somehow everyone has diff conditions that affect # of misfolding which icnrease prion disease chances.
Once PrPSc proteins activated
Once aggregates begins, the pace of making more misfolding increases. After some point, very small increase but becomes huge # of misfolding occurs.
Lower activation barrier,proteins refold into PrPSc rapidly like chaperonins
Familial prion disease - caused by what mutation? what type of pedigree?
Q36. If one parents display fCJD, what probability of their child diagnosis to fCJD?
Familial prion disease is point mutation affect protein folding. AA change makes big difference. Diff types of prion disease by having small mutation in prion gene. fCJD - AD pedigree. Many people in fam are affected and most likely every gen. Same like huntington's disease where patient shows symptoms later in their life.
50%. 2 chromosomes (chromosome 1+2) so one is given unless i say both have dominant condition.
AD; point mutation: 50%
Sickle cell cure and stuck in what
Sickle cel disease have hypoxia and necrosis (cell death) if blood supply low due to crescent shape RBC get stuck in capillaries. Sickle cell disease curable by cutting edge technology.
What technology would u use to make it happen? CRISPR genome editing. Good thing is blood cells (stem cells) easy to infuse. Blood stem cells include all type of cells including RBC. By thinking of treatment, blood cells easier to treat than brain. If jakob disease have a mutation and gray have a mutation, harder to infect brain.
CRISPR; capillaries
What techniques used to determine changes of protein structures?
X-ray crystallography or NMR or cryoEM. Each diff technique have pros and cons as some are good for small or some for big.
Amylodosis technology for treeatment
Amylodosis is genetic disease so it requires CRISPR genome editing and trying to reduce symptoms due to accumulation of amyloid (abnormal proteins).
Hb1Ac - glycation or glycosylation
Hb1Ac not used to determine the patient is diabetic bc most imp test is to test blood glucose. The tendency of last months of blood glucose levels can be observed/assessed by checking glycation.
glycation