experiment 6 (week 2): agarose gel electrophoresis Analysis of spinach DNA and restriction digest analysis of plasmid DNA
1/111
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
112 Terms
molecular cloning
recombining different pieces of DNA
-method that allows researchers to transfer a small portion of DNA, from one DNA molecule into a different DNA molecule and making numerous copes of the new hybrid molecule.
restriction enzymes
-the ability to cut out a piece of DNA is dependent upon a specialized group of enzymes found in bacteria
what do restriction enzymes cut
restriction enzymes are proteins that cut the sugar phosphate backbone of DNA only when a specific sequence of nucleotides is present.
recognition sequence
A specific sequence of nucleotides at which a restriction enzyme cleaves a DNA molecule
restriction enzyme Xhol
only cuts between nucleotides C and T of the following sequence
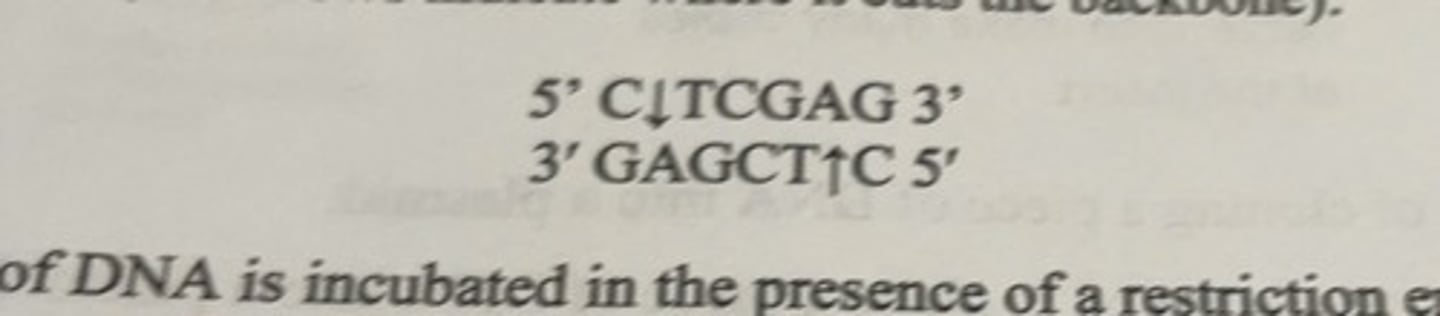
what happens when a restriction enzyme cuts
when a piece of DNA is incubated in the presence of a restriction enzyme, the enzyme will cut wherever it finds its recognition sequence and a series of fragments will be produced.
what does the size of the fragments depend on
the specific sequence of the DNA and the restriction enzyme used.
digestion
-incubation and cutting process
-it is the precise control over the restriction digest fragments that enables experimenters to assemble different pieces of DNA from different DNA molecules into a new DNA molecule with the desired nucleotide sequence.
DNA molecules cut with the same restriction enzymes
the digestion will produce identical ends on both molecules that can then base pair together allowing the 2 molecules to be joined together.
plasmids structure
double stranded
circular
DNA molecules
what can plasmids do
can make copies of themselves inside bacteria, independent of the copying of the genomic DNA during cell replication
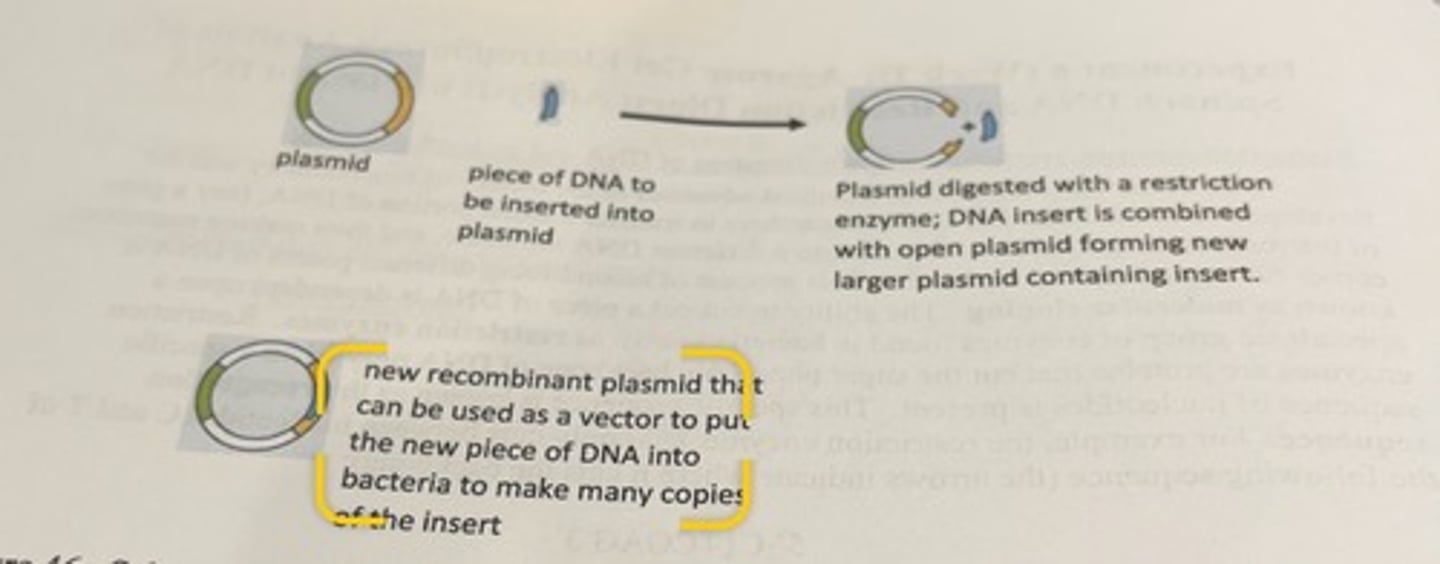
description of plasmid process
a plasmid is digested with a particular restriction enzyme so that the plasmid is cut open in a very particular way, then a new independent piece of DNA that contains the information that interests the researcher is combined with the opened plasmid, and then the plasmid is sealed using specific enzymes (ligases) creating a new larger plasmid.
what can the new recombinant plasmid do
-can be inserted into bacteria which can make numerous copies of the new plasmid.
-these new plasmids are then purified out of the bacteria and used for the next stage.
confirming the cloning process has worked
-if attempting to clone a piece of DNA into a plasmid, need to verify cloning process has worked
-can do this by digesting the new plasmid with restriction enzymes and then run the products of the digestion reaction on an agarose gel to see if you get pieces of the appropriate sizes.
-this analysis requires estimation of the sizes of digested DNA, we need a method to determine DNA size (agarose gel electrophoresis)
analyzing plasmid DNA by restriction digest analysis
selective restriction enzyme digestion to determine if the intended recombination between the DNA insert and plasmid has taken place.
scenario of analyzing DNA by restriction digest analysis
-plasmid A (5500 bp) has 2 restriction sites (R21 and RS2).
-they are cut by 2 different restriction enzymes (RE1 and RE2).
-the desired DNA insert is 850 bp (B)
-you want to recombine them to form a new larger plasmid (C).

2 possible outcomes
1. the reaction works and. you have a new larger plasmid with the insert placed between the 2 restriction sites (C)
2. the reaction failed, and you only get the original plasmid back (A).
which enzyme you use matters
-if you digest the 2 different possible plasmids with restriction enzymes 1 and/or 2, you get different results depending on which plasmid has been digested.
if you digest the original plasmid with 1 of the 2 restriction enzymes
you get a linear piece of DNA that is 5500 base pairs.
if you digest it with both RE1 and RE2
-will still obtain a linear piece of DNA that is about 5500 bp in length (lose the little piece of DNA between the 2 sites).
if you digest the recombinant plasmid with the insert (C) with one of the 2 enzymes
-will get a linear piece of DNA that is 6350 base pairs in length
if you digest C with both restriction enzymes
-you will get one piece of linear DNA that is 5500 bp
and
-you will get a second piece of DNA that is the size of the insert (850 bp)
what is the next step
to determine the actual sizes of the DNA pieces after digestion
what are the easiest ways to determine the sizes of the digestion reaction products
-electrophoresis
electrophoresis
-use agarose gel that separates DNA based upon the sizes of DNA
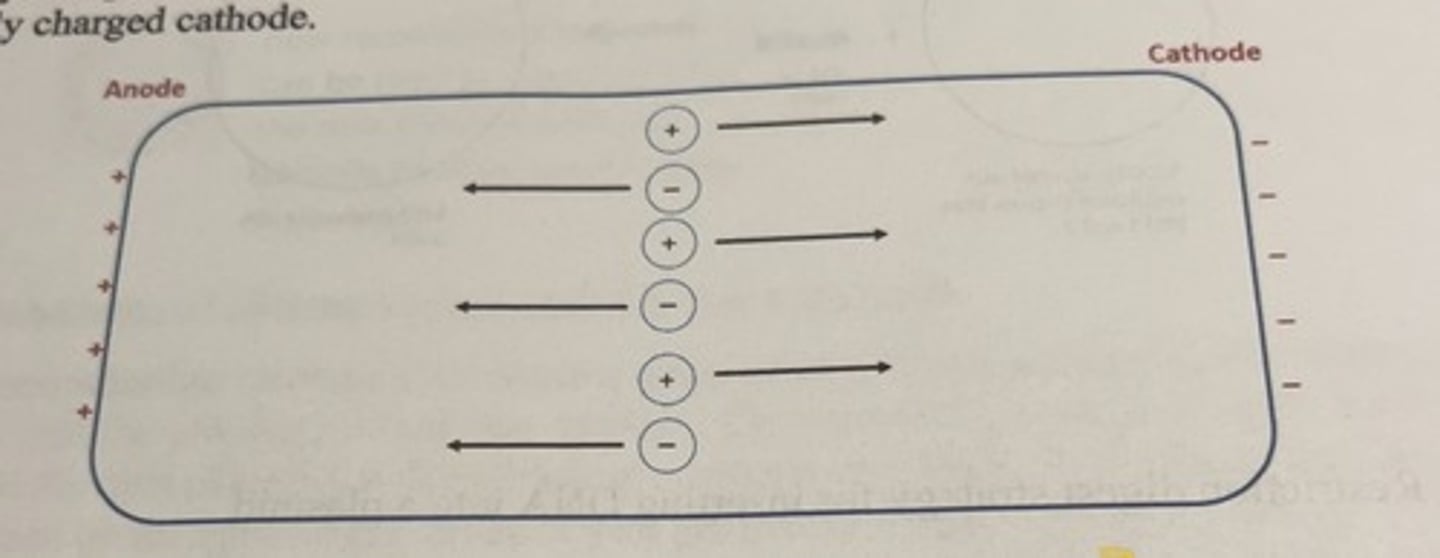
When charged molecules are placed in solution and exposed to an electrical field
the molecules will have the tendency to migrate to the pole opposite of their charge.
-negatively charged molecules migrate towards the positively charged anode
-positively charged molecules will migrate towards the negatively charged cathode.
electrophoretic techniques
many techniques used in biochemistry that exploit separating charged molecules.
what occurs during electrophoresis
-examination of the movement of charged particles through solution under the influence of an electric field
-when you apply a DNA sample to one end of a gel and activate the voltage, the negatively charged DNA will begin to migrate towards the positively charged pole at the opposite end of the gel.
one of the most useful attributes of electrophoresis
-the speed at which a molecule moves towards the oppositely charged pole can be different for different molecules.
migration velocity
v = velocity of the charged particle
E = strength of the electric field to which the charged molecule is exposed
q = net charge on the particle
f = frictional coefficient of the particle as it moves through the solution.
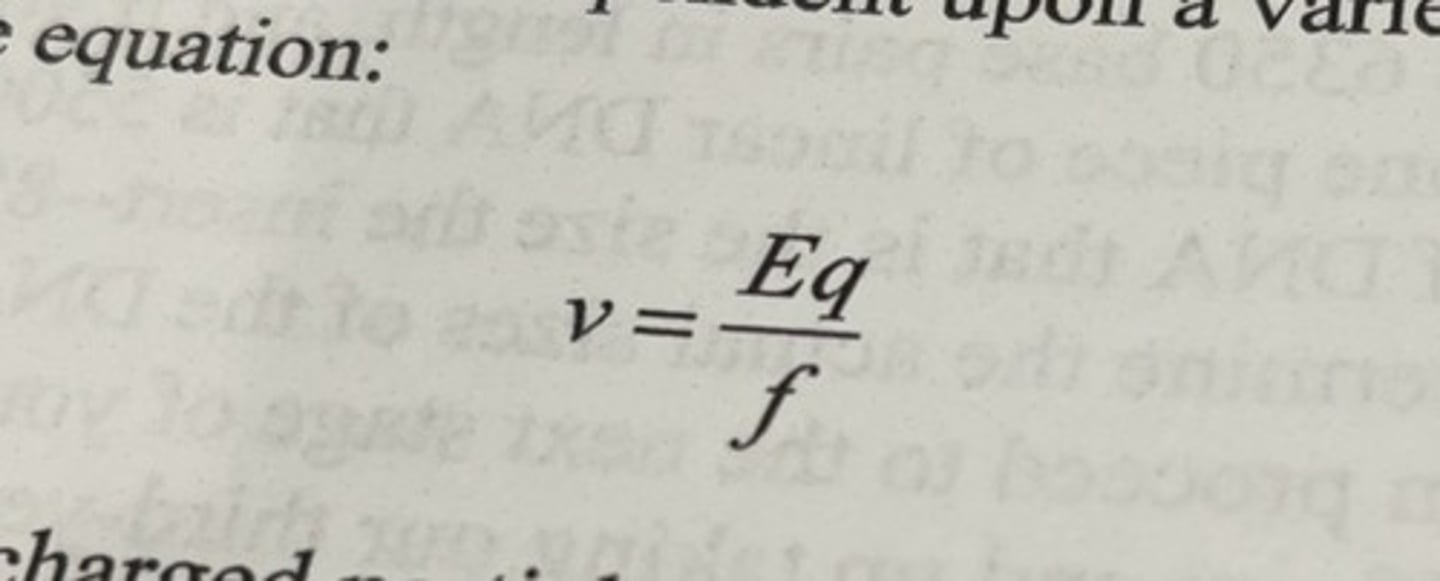
what happens when you apply an electric field to charged molecules
they will start to accelerate towards the pole of the opposite charge
how fast a molecule accelerates depends on
both the strength of the field and how much charge (q) the molecule carries
viscous drag
-as molecule moves faster, viscous drag increases from moving molecule through solvent..
what is this drag influenced by
size and shape of the molecule
how does the viscous drag act
-it is a force of opposite direction to that of the migrating particle
-the size and shape of the molecule is represented by the frictional coefficient (f)
steady state velocity
-almost immediately after the voltage is applied and maintained at a constant level, the force accelerating the molecule forward (Eq) is equalized by the force f, decelerating it.
-steady state velocity or migration speed through the solvent is attained.
-this steady state velocity is denoted by the term v from the above equation.
E for molecules
-in any specific electrical field, E is the same for all molecules in that field
-the migration velocity of any particular molecule will depend upon its q/f ratio
charge to mass ratio
-based on q/f ratio
-this term is a mistake
-the magnitude of f is extremely complex, and is determined in part by the size and shape of the charged molecule.
structure of DNA at neutral pH
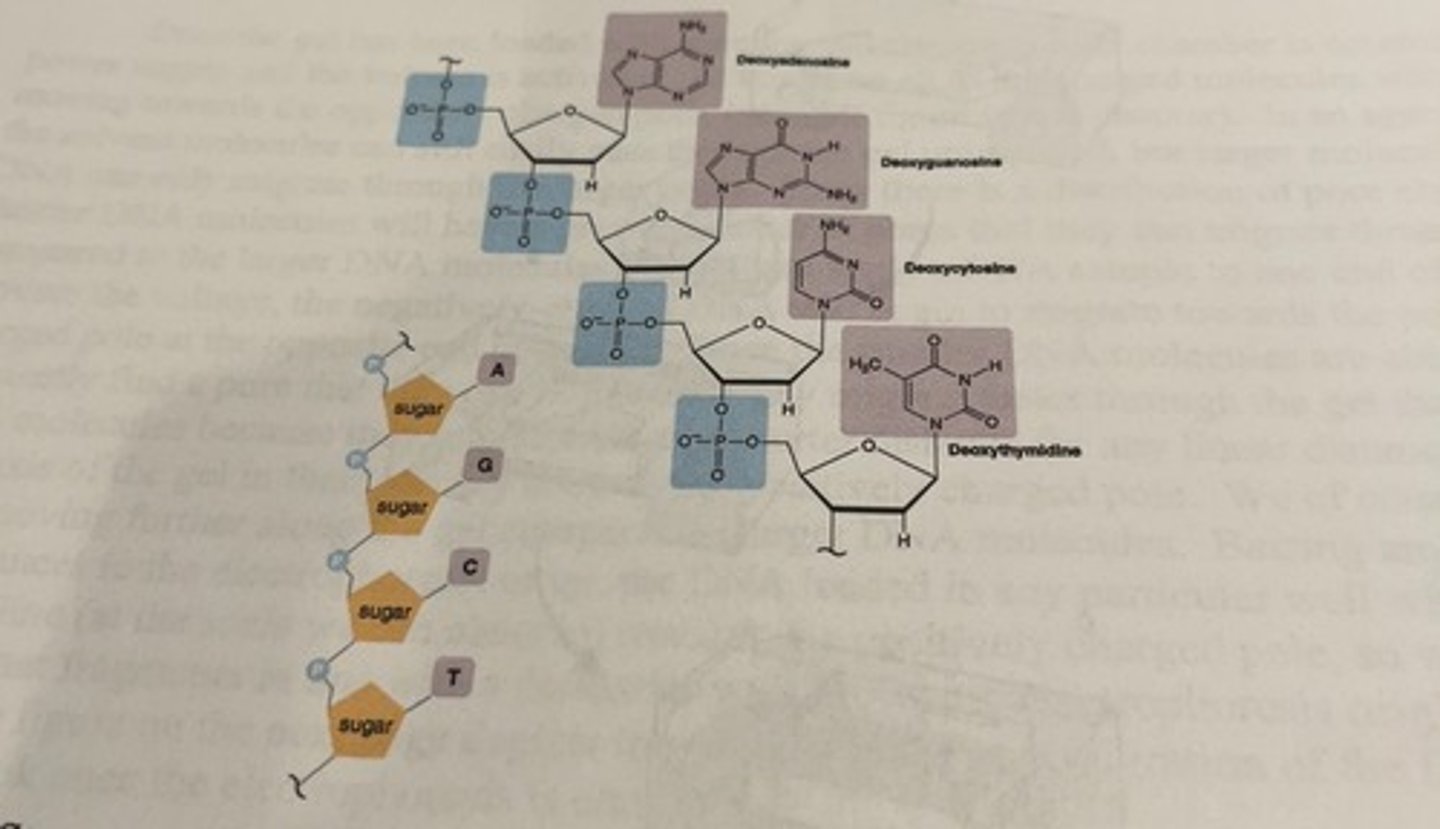
charge of phosphate groups
-the phosphate groups impart an overall negative charge to the molecule which means that it will migrate towards the positively charged pole when voltage is applied
-the amount of negative charge on a DNA molecule is proportional to its length (since there is a negative charge for each nucleotide)
q/f ratio for DNA molecules
DNA molecules of different length have a similar q/f ratio
-in theory, migrate at a similar steady state velocity
-DNA molecules regardless of their size would migrate at the same speed in electrophoresis.
-problem: how is it useful to the researcher if all the DNA molecules behave the same.
solution
need to use special materials that do allow for a size-based separation
gel
to separate DNA molecules of different lengths, we need to perform the electrophoresis in a porous filled mechanical support that is called a gel.
what material is gel made of
any material that forms a sieving matrix that has minimal interaction with the molecules one is trying to separate.
what are gels made of when using DNA
agarose
what is agarose
-a linear polysaccharide that forms a mesh network in aqueous solution when it is melted in solution and allowed to resolidify.
what size molecules do we use agarose for
typically used for the electrophoretic separation of large biomolecules such as nucleic acids and also proteins with a mass greater than 250 kDa.
the mesh network
formed by solidication of molten agarose creates a gelatinous-like substance that is filled with pores of varying sizes.
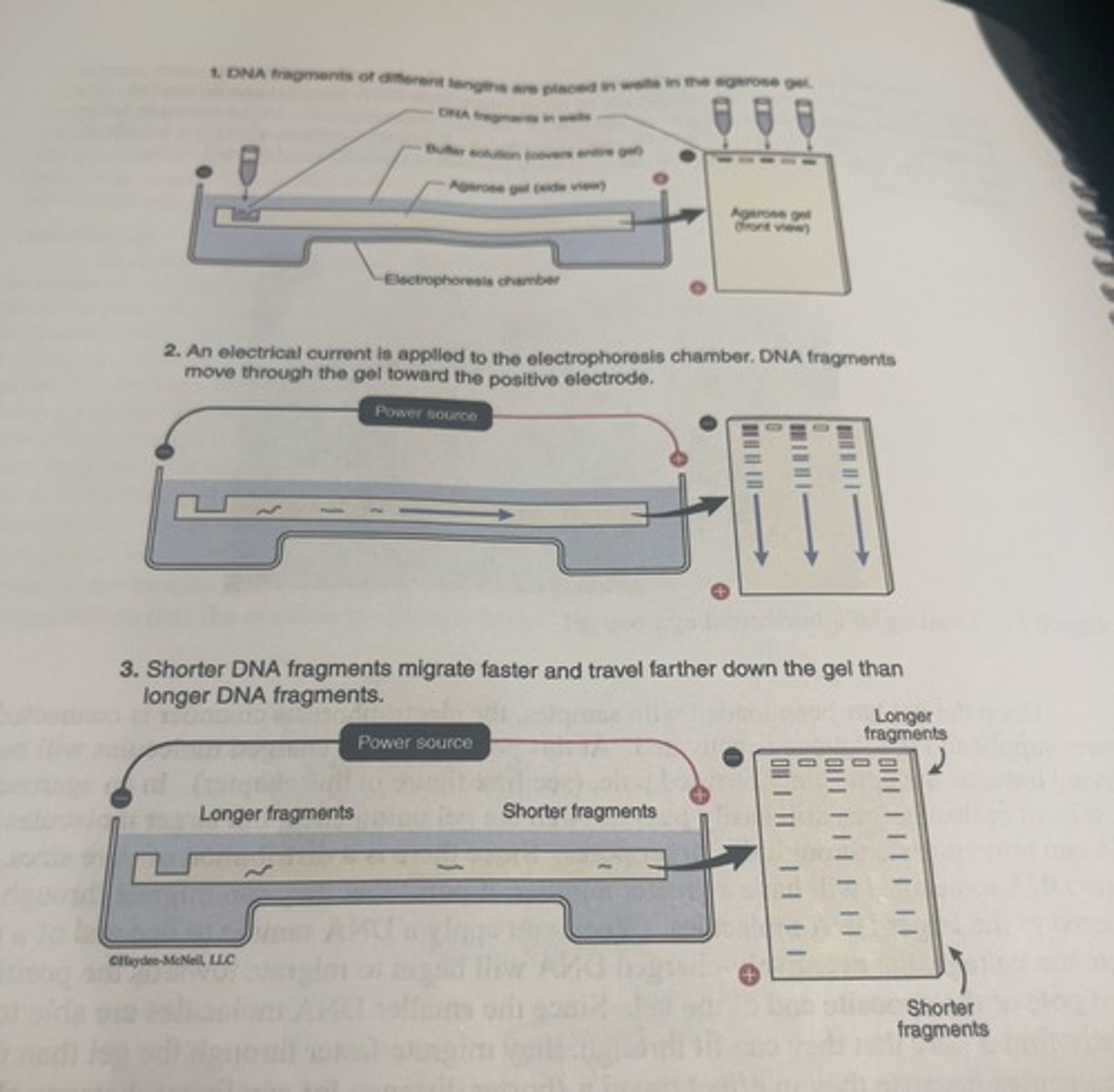
casting of a horizontal agarose gel
-a gel casting tray is placed in a larger electrophoresis chamber (A) in such a way as to minimize leaking, and then a comb is placed in the tray followed by the addition of the molten agarose solution (B). the gel hardens in the casting tray and then the tray is rotated 90 degrees so that the comb is positioned at the negative pole of the electrophoresis chamber (C)

what do you do with the comb
-once gel has hardened, remove comb.
-where the teeth of the comb were in the gel will now be a series of evenly spaced holes into which DNA samples can be loaded.
multiple cones
multiple cones can be used so that DNA can be loaded at different distances along the length of the gel
-the gels we use in the lab only have a comb at one end.

once the gel has been loaded with samples
-the electrophoresis chamber is connected to a power supply and the voltage is activated.
-at this point, all the charged molecules will begin moving towards the oppositely charged pole.
different molecules running through agarose gel
-the solvent molecules can still easily pass through the gel unimpeded
-larger molecules like DNA can only migrate through the larger pores.
pore sizes in agarose gel
-there is a distribution of pore sizes
shorter DNA molecules in agarose gel
shorter DNA molecules will have a greater number of pores that they can migrate through compared to larger DNA molecules.
-since the smaller DNA molecules are able to more frequently find a pore that they can fit through, they migrate faster through the gel than the larger DNA molecules because they in effect travel a shorter distance for any linear distance along the long axis of the gel in their journey towards the positively charged pole.
-they will see them moving further along the gel compared to larger DNA molecules.
DNA loading in a well
-barring any external disturbances to electrophoresis setup, the DNA loaded in any particular well will move in a straight line towards the positively charged pole, so we can safely assume that fragments in line with a particular well following electrophoresis originated in that well.
how to get the desired agarose concentration in the buffer
-they are prepared by adding enough agarose to an appropriate buffer
how do you change agarose pore size
one can vary the average pore size of the gel by changing the concentration of the agarose.
higher agarose concentrations
lower average pore size
what is the typical agarose concentration
typically the concentration of the agarose is between 0.5 and 3.0% agarose to buffer on a mass/mass basis, and the concentration you choose depends upon the expected ranges of sizes of DNA that you are trying to separate.

gels below 0.5%
tend to fall apart
gels greater than 3.0%
one is often advised to switch to polyacrylamide gel instead.
how to calculate the mass of agarose in grams that is required to prepare a gel
volume of gel desired (in mL) X 0.01 X agarose %
-then you will add this calculated mass of the agarose to mL (that you plugged in) of buffer.
solubility of agarose
-it is not very soluble at room temperature, but when the buffer is heated to near boiling temperatures, the agarose will dissolve.
what happens to agarose when the solution is allowed to cool
the agarose solidifies, forming the gel.
how long do you heat agarose gel for
30-40 seconds
-greater volumes will require longer heating
-you do it in a microwave until it almost boils.
this molten agarose solution is then transferred to a casting chamber and a comb is added at one end so that wells for loading the samples are formed when the solution cools.
when is the buffer poured in
after the gel solidifies, buffer is poured into the casting chamber to completely submerge the gel and then the comb is removed and samples are added to individual wells.
what is the problem if determining DNA speeds
-there is no universal speed at which DNA moves in a gel because every gel has different concentrations of agarose, is run at different voltages, and will vary in buffer composition and a host of other factors that can slightly affect the migration speed of the DNA.
solution
-a DNA sample that contains various pieces of DNA of known sizes is also loaded on the same gel.
DNA ladder enables you to
calibrate the gel because you will be able to see exactly how far different known sizes of DNA will move on the gel under conditions essentially identical to those of your samples.
gel running conditions
-gels are typically run at 6-10 V/cm
-this means that if gel was 10cm in length, you would run this between 60-100 V.
-gels that are run at higher voltages do allow the DNA to migrate faster
setting the voltage too high
-can often result in overheating which can lead to variety of artefacts including smearing of the DNA.
is DNA readily visible on the gel
no
how do you see the DNA
-chemicals that are easily detected and that interact only with the DNA are used.
-most of the commonly used DNA dyes are based upon fluorescence (allows for high sensitivity).
historically, what was the dye used for visualization of DNA
ethidium bromide.
ethidium bromide
dye that stacks in between the nitrogenous bases and fluoresces at a visible wavelength when irradiated with UV light
-this dye is a potential carcinogen.
-disposal of gels that contain this dye are subject to stringent regulations
-use of this dye has diminished. safer and more expensive dyes are now routinely used.
examples of new dyes
Red safe
SYBER safe
HydraGreen
SYBR gold
capabilities of DNA binding dyes
different DNA dyes have different capabilities for detecting DNA
SYBR gold
the most sensitive dye
-can detect <50 pg of DNA in a single band.
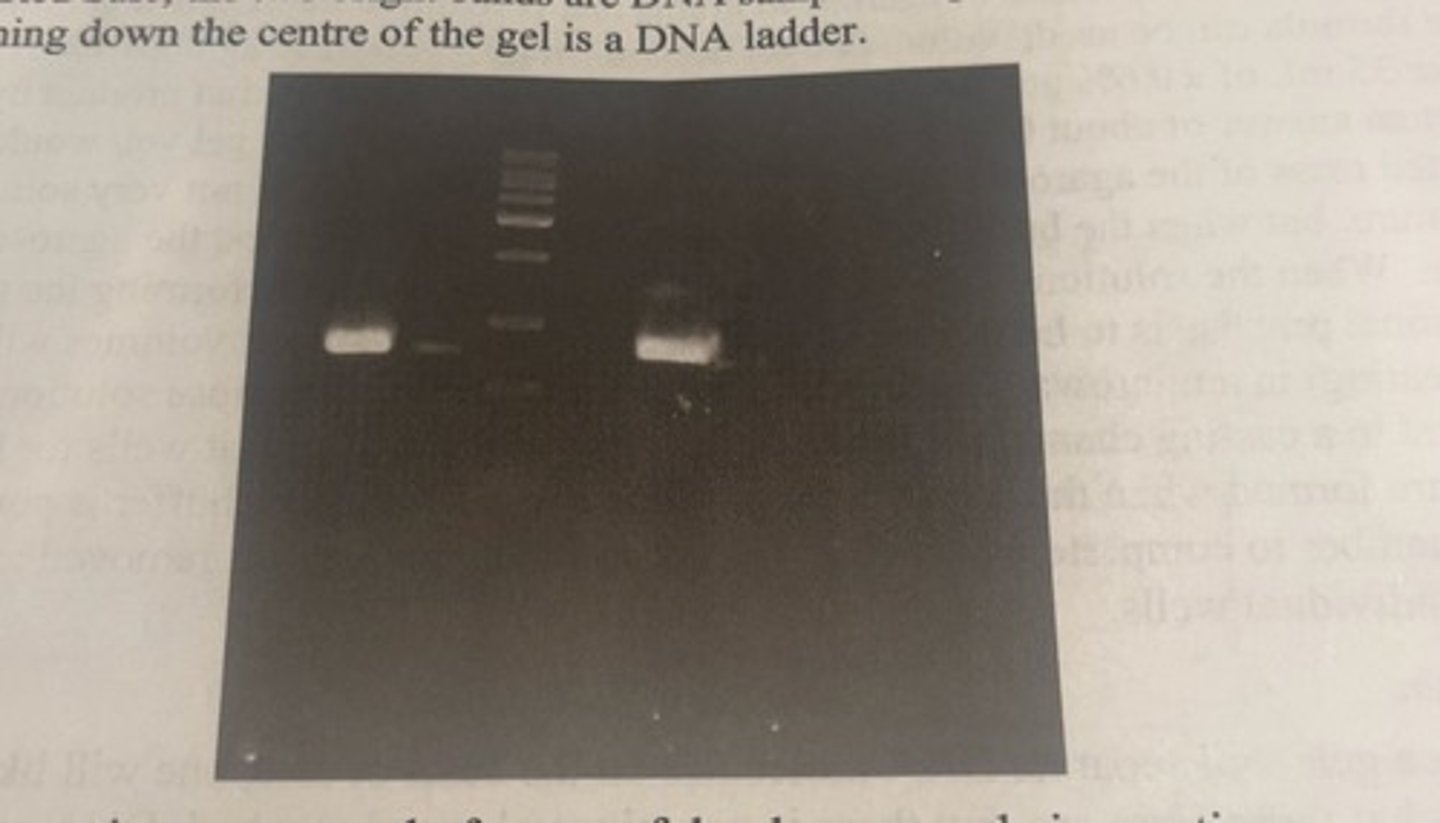
agarose gel stained with red safe
-the 2 bright bands are DNA samples amplified by PCR
-the series of bands running down the centre of the gel is a DNA ladder.
by analyzing DNA by gel electrophoresis, what can we get from this
-analyze the digestion of products of a restriction enzyme digestion of plasmid DNA for evaluating cloning success.
-assessing purified DNA quality
-confirming PCR success
-isolating specific DNA molecules
DNA quality
-the quality of extracted DNA can be assessed from several perspectives, including purity and the size of the extracted DNA fragments.
how is purity most readily determined
by looking at the absorbance of the DNA sample at 260 and 280 nm which can reveal the extent of protein contamination
what is pure DNA
free of protein
quality of degraded DNA
-even if a DNA sample is pure, it may not be high quality if it is extensively degraded.
fragmentation of eukaryotic genomic DNA
-inevitable as the removal of histones and other stabilizing proteins leaves the DNA very fragile, and it invariably degrades into smaller pieces.
is this problematic?
no, as long as the average pieces are relatively large.
-larger than 10 kilobases because pieces of this size will leave numerous copies of individual genes and regulatory DNA intact, and therefore the DNA sample can still be used for whatever purposes it was originally purified.
if DNA were smashed into much smaller fragments
-it would be much less likely to contain intact pieces of desired sequences
-unlikely to generate good results in the subsequent stages of a longer experimental protocol.
determining if DNA has been overly physically degraded
-since size is one of the properties of DNA that can be readily ascertained using a gel, running the sample on an agarose gel is a quick and east way to determine if the DNA has been overly physically degraded and therefore can also be used to evaluate the quality of the purified DNA.
why does purified DNA always show a smear
purified eukaryotic DNA will always show a smear on the gel because of the random nature of the fragmentation which mean you could have numerous pieces of DNA of similar, but not identical sizes.
-the similarity of the DNA sizes will cause them to run at similar but not identical speeds which occurs as a smear.
the key for quality assessment
what size range does the smear cover
primary purpose of PCR
to make millions or even billions of copies of a specific piece of DNA.
confirming PCR success
-the conditions necessary for PCR to succeed are well understood and usually successful, but because the different pieces of DNA that you might want to copy require slightly different reaction conditions, it is always important to verify that the reaction has worked.
-since the researcher will usually know the size of the DNA they want to make copies of, running a small sample of the PCR product on a gel allows the researcher to confirm success as there should be a single bright band of the expected size.
DNA isolation
agarose gel electrophoresis can also be used to purify a specific piece of DNA from other DNA molecules.
-this method only works if your desired piece of DNA is bigger or smaller than other DNA that might happen to be in your sample.
-to purify it, you run it on the gel which separates it from the other DNA molecules and then you can literally cut out the piece of agarose gel that contains the DNA that you want with a scalpel.
-the DNA in that small piece of gel can then be purified from the gel and gel buffers using spin column based technology that is very similar to what is used to purify DNA from cells.
-since ideally there will be only one type of DNA in the cut of agarose gel, you will end up with a highly purified single type of DNA.
what restriction enzymes were used in lab 6 week 2
BamHI and NdeI
what did you do after digestion in lab 6 week 2
run the digested plasmid DNA on the same gel as your spinach DNA.
-the E. coli cells that were provided to students in the 1st week of this experiment either had a plasmid with an insert or a plasmid lacking the insert.
-by analyzing results of the digestion reaction, you can determine which plasmids did and did not contain the insert.
what % agarose did we use
0.6%
what buffer did we use with the agarose
TRIS acetate ethylenediaminetetraacetic acid (TAE) buffer
where do you place the comb
as near as possible to the end of the casting tray
-the comb sits in slots cut into the walls of the casting tray

how long does it take for the gel to solidify
20 minutes