Genetics E2- Neuro & Muscular Dystrophies
1/71
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
72 Terms
What autosomal dominant condition is a fatal, progressive neurodegenerative disease of the CNS?
Huntington’s Disease
What causes Huntington’s disease / chorea?
Expansion of CAG trinucleotide in the HTT gene on 4p16.3, which encodes huntingtin
When does Huntington’s disease most commonly onset?
30-60 y/o
What symptoms are seen in Huntington’s?
Progressive chorea & insidious impairment of intellectual function (psychiatric → dementia)
What are brief, abrupt, involuntary, non stereotyped movements that involve the face, trunk, & limbs?
*key feature of Huntington’s disease
Chorea
What is the treatment for Huntington’s disease?
Chorea: VAMT2 inhibitors (use antipsychotics if depression)
Depression: SSRIs
*no disease-modifying therapies available
What drugs are VMAT inhibitors?
Tetrabenazine, duetetrabenazine
How do VMAT2 inhibitors treat chorea that interferes with function?
Inhibit presynaptic DA, thereby inhibiting release of DA at the synapse
When should VAMT2 inhibitors be avoided in the treatment of Huntington’s?
If depression is present → use antipsychotics instead
What does CADASIL stand for?
Cerebral autosomal dominant arteriopathy w/ subcortical infarcts & leukoencephalopathy
What autosomal dominant condition is a microangiopathy affecting mainly the brain, and is also the MC inherited cause of stroke & vascular dementia?
CADASIL
What causes CADASIL?
NOTCH3 gene on 19p13.2 → missense variants (MC) or cysteine-altering variants (causes vascular lesions in small arts&caps)
The following clinical features are seen with what condition?
Migraine w/ aura (often initial manifestation)
Acute reversible encephalopathy
Ischemic episodes (MC manifestation)
Cognitive impairment & dementia
Psychiatric disturbances
CADASIL
What is the typical clinical course of CADASIL?
Age 30: migraine w/ aura
40-60: TIA, ischemic strokes, mood d/o
50-60: dementia
~60: gait difficulty
What are the 2 major typical findings of CADASIL on an MRI?
Small, circumscribed regions isointense to CSF on T1 & T2 weighted images
Less well demarcated T2 hyperdensities/T1 hypodensities clearly distinct from CSF
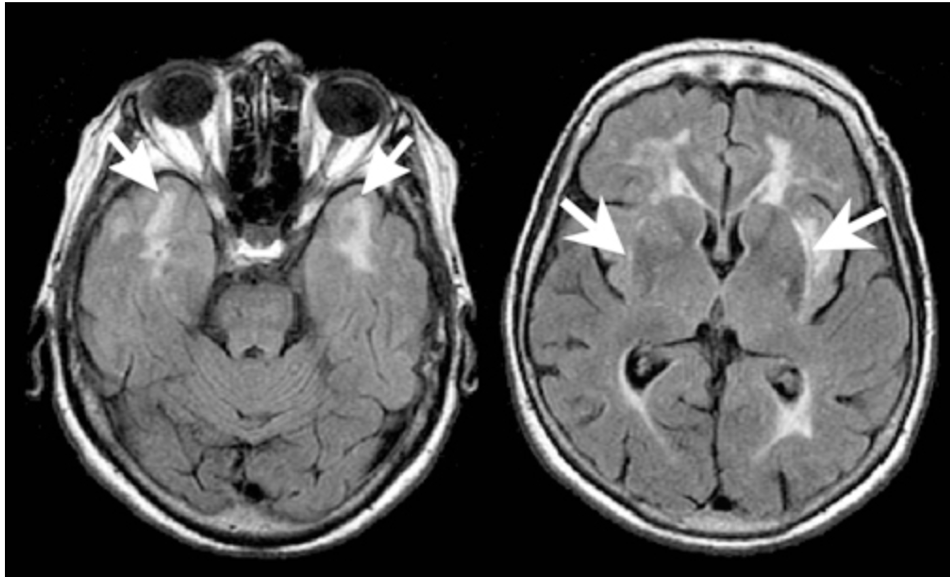
What is the treatment for CADASIL?
CVA: tx acute stroke, HTN, hypercholesterolemia; low dose ASA for prevention
Dementia: donepezil
Migraine: NSAIDs, avoid triptans
What are causes of familial early onset Alzheimer’s disease?
PSEN1 chromosome 14q (MC, ~43 y/o onset)
PSEN2 chromosome 1q,
APP chromosome 21q
What inheritance pattern does familial early onset alzheimer’s disease follow?
Autosomal dominant; very high penetrance (PSEN1/APP > PSEN2)
What clinical manifestations are seen in familial early onset AD?
Memory impairment, poor judgement, agitation, withdrawal, confusion, language difficulties, occasionally parkinsonian features & seizures
When should genetic testing for familial early onset AD be done?
If symptomatic or if an asymptomatic pt has a FHx suggesting inheritance
What are progressive conditions characterized by motor incoordination d/t dysfunction of the cerebellum & it’s connections, often accompanied by wide-based gait, dysarthria, & nystagmus?
*can be AD, AR, X-linked, or mitochondrial inheritance
Hereditary ataxias
Which hereditary ataxia is autosomal dominant & characterized by intermittent periods of unsteady gait, lasting several hours, with nystagmus and dysarthria?
Episodic ataxias
Which condition is an autosomal recessive progressive ataxia of all limbs and is the MC hereditary ataxia?
Friedreich ataxia
What genetic mutation is involved in Friedreich ataxia?
Loss-of-function mutation on frataxin (FXN) gene on 9q13
What clinical features are seen in Friedreich ataxia?
*Dx’d w/ genetic testing
Progressive ataxia of all limbs, neurological dysfunction, cardiomyopathy, DM, life expectancy ~ 40-50 yrs (some can live beyond 60s)
What is another name for Charcot-marie-tooth disease (CMT)?
Multiple hereditary sensory & motor neuropathies (HSMN)
What spectrum of disorders are caused by mutations in genes whose protein products are expressed in myelin, gap junctions, or axonal structures w/in peripheral nerves?
*characterized by slowly progressive distal muscle weakness & wasting - “stork legs”
CMT
The following clinical features are associated with what condition?
Distal weakness & atrophy of intrinsic muscles (MC initial sx) -
foot drop
pes cavus
stork legs
hammer toes
sensory symptoms (less prominent)
CMT
How is CMT diagnosed?
EMG → confirm w/ genetic testing
What is another name for amyotrophic lateral sclerosis (ALS)?
Lou Gehrig disease
What presents with focal, asymmetric weakness in the extremities & bulbar signs such as dysphagia or dysarthria?
ALS
What is the average age of onset & survival of ALS?
56 y/o; 1-5 years after dx
What kind of inheritance pattern does familial ALS have?
MC autosomal dominant but can be autosomal recessive or X-linked
*but most cases are sporadic not familial (90-95%)
What genes are MC implicated in familial ALS?
C9ORF72: assoc w/ high risk FTD
SOD1: ranges from rapid progression (< 2 yrs) to slow (10-20 yrs)
also TARDBP & FUS
How does ALS affect the nervous system?
Degeneration & death of motor neurons → gliosis replaces lost neurons → spinal cord becomes atrophic → ventral roots become thin → affected muscles show denervation atrophy, unable to contract
What LMN signs are seen in ALS?
Weakness, fasciculations, muscular atrophy, hypotonia
What UMN signs are seen in ALS?
Hypertonia, spasticity, inc DTRs, +babinski, pseudobulbar affect (inappropriate laughing, crying, forced yawning)
What is the Gold Coast diagnostic criteria for ALS?
Progressive motor impairment
Presence of either UMN & LMN sx in atleast 1 limb/body segment OR progressive LMN sx in atleast 2 body segments
Absence of electrophysiologic, neuroimaging, and pathological evidence of other diseases
What are the treatment options for ALS?
Riluzole: slows progression, modest survival
Edaravone: slow functional decline in early stages
Tofersen: targets SOD1 gene mutations
What is another name for Neurofibromatosis type 1 (NF1)?
*MC type of neurofibromatosis
Von Recklinghausen disease
What is a benign, unencapsulated tumor composed of a mixture of cell types from the nerve sheath (Schwann cells, fibroblasts, mast cells)?
Neurofibroma

How many features of the diagnostic criteria must be present for a diagnosis of NF1?
cafe au lait macules, neurofibromas, Crowe sign, optic pathway glioma, linch nodules or choroidal abnormalities, distinctive osseous lesion, heterozygous variant
2 or more
What is axillary or inguinal freckling, seen in NF1?
Crowe sign
What condition is due to a mutation in the NF1 gene on 17q11.2, leading to a lack of tumor suppression which results in the growth of neurofibromas?
NF1
What inheritance pattern does NF1 follow?
Autosomal dominant
*50% have FHX; 50% are de novo mutations
What is a de novo mutation?
Spontaneous genetic alteration that appears for the first time in a family, not inherited from either parent
What is the treatment for NF1?
Lifelong surveillance, targeted therapies (selumetinib for plexiform neurofibromas), & surgery for symptomatic neurofibromas
What is the prognosis of NF1?
Life expectancy reduced 8-15 yrs, median survival ~ 70 y/o
What condition is characterized by the following clinical findings?
Hallmark: BL VS on vestibular portion CN VIII → progressive hearing loss, tinnitus, & balance dysfunction
Ocular: juvenile posterior subcapsular or cortical cataracts, epiretinal membranes
Cutaneous: skin schwannomas, cafe au lait macules
NF2-SWN
What other tumors are commonly associated with NF2?
Multiple meningiomas (cranial & spinal), schwannomas on other nerves, & spinal ependymomas (intramedullary)
When does NF2 typically onset?
Late teens-early 20s; most develop BL VS by age 30
How is NF2 diagnosed?
Manchester criteria or identification of pathogenic variant

What condition is caused by a mutation of the NF2 gene on chromosome 22, disrupting merlin activity & leading to the formation of schwannomas?
NF2-SWN
What inheritance pattern does NF2-SWN follow?
Autosomal dominant
What is the management for NF2-SWN?
Annual MRI, spinal MRI q 2-3 yrs, surgery, Bevacizumab
*progressive condition, median survival 60-70s+
What monoclonal antibody is an angiogenesis inhibitor that has been shown to reduce the size of vestibular schwannomas & some spinal tumors?
*used to tx NF2-SWN
Bevacizumab
What protein product is involved in NF1?
Neurofibromin (tumor suppressor, widely expressed)
What protein product is involved in NF2-SWN?
Merlin (aka Schwannomin; cell membrane related protein that acts as tumor suppressor)
What conditions are prominent X-linked recessive dystrophinopathies that are caused by mutations of the dystrophin gene on Xp21.2 & occur primarily in males?
DMD & BMD
Which muscular dystrophy involves absent/deficient dystrophin?
Duchenne (DMD)
Which muscular dystrophy involves abnormal/reduced dystrophin?
Becker (BMD)
Which muscular dystrophy presents with a more severe clinical phenotype & earlier onset?
Duchenne (DMD)
What is a rod shaped cytoplasmic protein that acts as an anchor connecting each muscle cell’s cytoskeleton with the extracellular matrix?
*deficiency causes muscles fibers to be destroyed → replaced by fat & fibrous tissue → DMD/BMD
Dystrophin
What clinical presentation is seen with DMD & BMD?
*more severe in DMD
Progressive muscle weakness, awkward gait, inability to run, gower’s sign, pseudo hypertrophy of calves
What sign seen in DMD/BMD is characterized by difficulty rising from the floor, achieved only by pushing on or climbing up legs?
Gower’s sign
What is the prognosis of DMD?
Boys present 2-4 y.o; wheelchair dependence by early-mid teens, median survival 30s; rarely reproduce
What are the chances of a son of a carrier mother inheriting a pathogenic allele for DMD (X-linked recessive)?
50%
What is the chance of a daughter of a carrier mother & unaffected father of becoming a carrier of DMD (X-linked recessive)?
50%
What gene is affected in DMD?
DMD gene on Xp21.2
What is the prognosis for BMD?
Ambulation maintained into mid-adulthood (40s-50s) or longer; life expectancy into middle age (50s-60s) or beyond
*cardiac health is key determinant
What is the diagnosis for DMD & BMD?
Gold standard: genetic testing for DMD mutations; if inconclusive → muscle bx
Early indicator: elevated CK in early childhood
What is the treatment for DMD & BMD?
Glucocorticoids to slow decline of motor & pulm function
Novel mutation specific therapies (DMD) to restore dystrophin: exon skipping, stop-codon read-through, gene therapy