Orgo Final Study Guide (CH1-12)
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
88 Terms
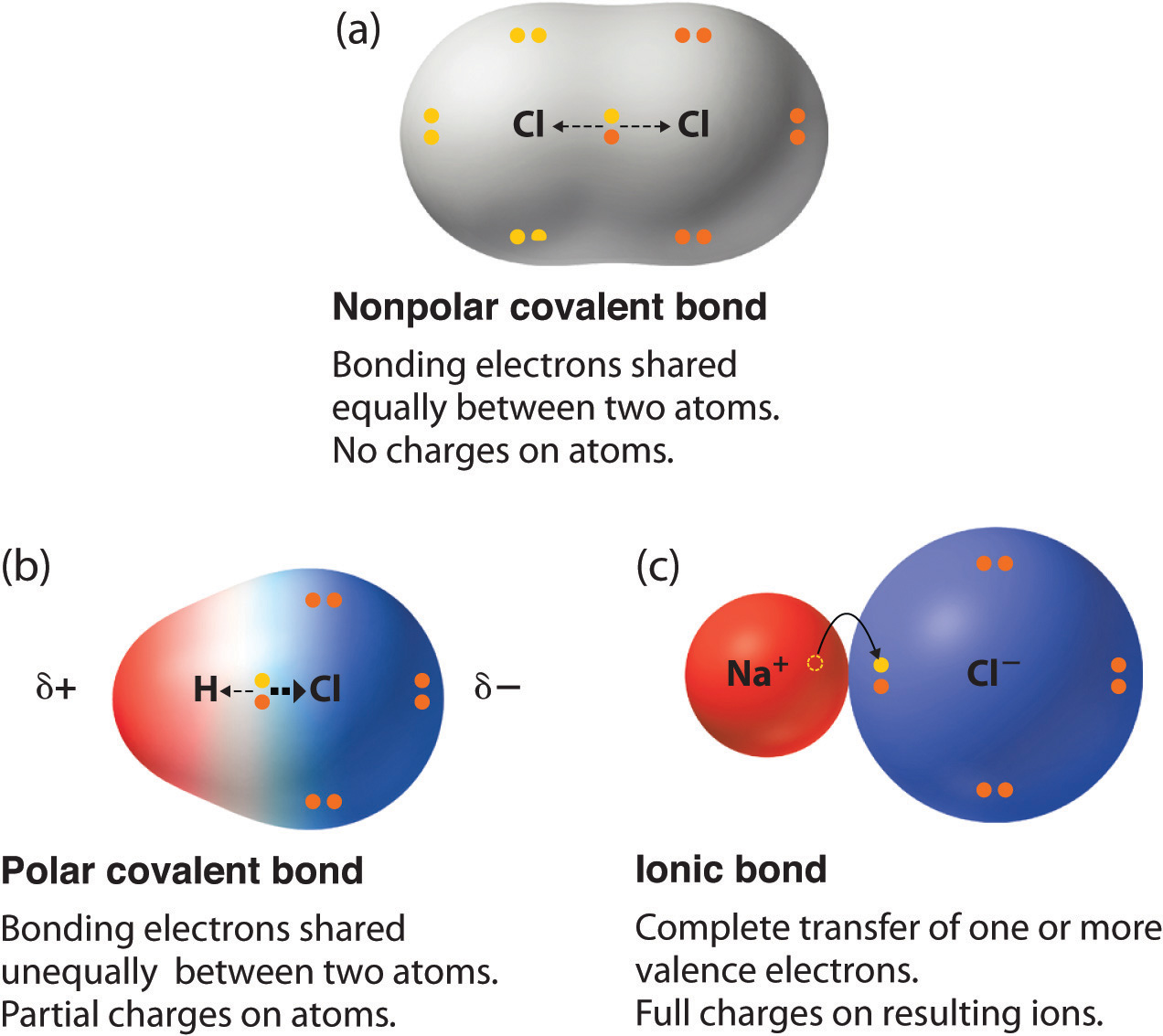
Recognize and explain the difference between ionic, covalent, and polar covalent bonds.
Ionic bonds occur when electrons are completely transferred from one atom to another, creating oppositely charged ions (e.g., NaCl).
Covalent bonds involve the sharing of electrons between atoms (e.g., H₂, CH₄).
Polar covalent bonds are a middle ground, where electrons are shared unequally due to differences in electronegativity (e.g., H₂O, HF).
Draw Lewis structures for compounds containing C, N, O, H, F, Cl, Br, and I.
Determine the total number of valence electrons by adding up electrons from all atoms.
Place single bonds between atoms and complete octets (or duets for H) with lone pairs.
Use double or triple bonds if necessary to satisfy octets.
Check formal charges to ensure the most stable structure.

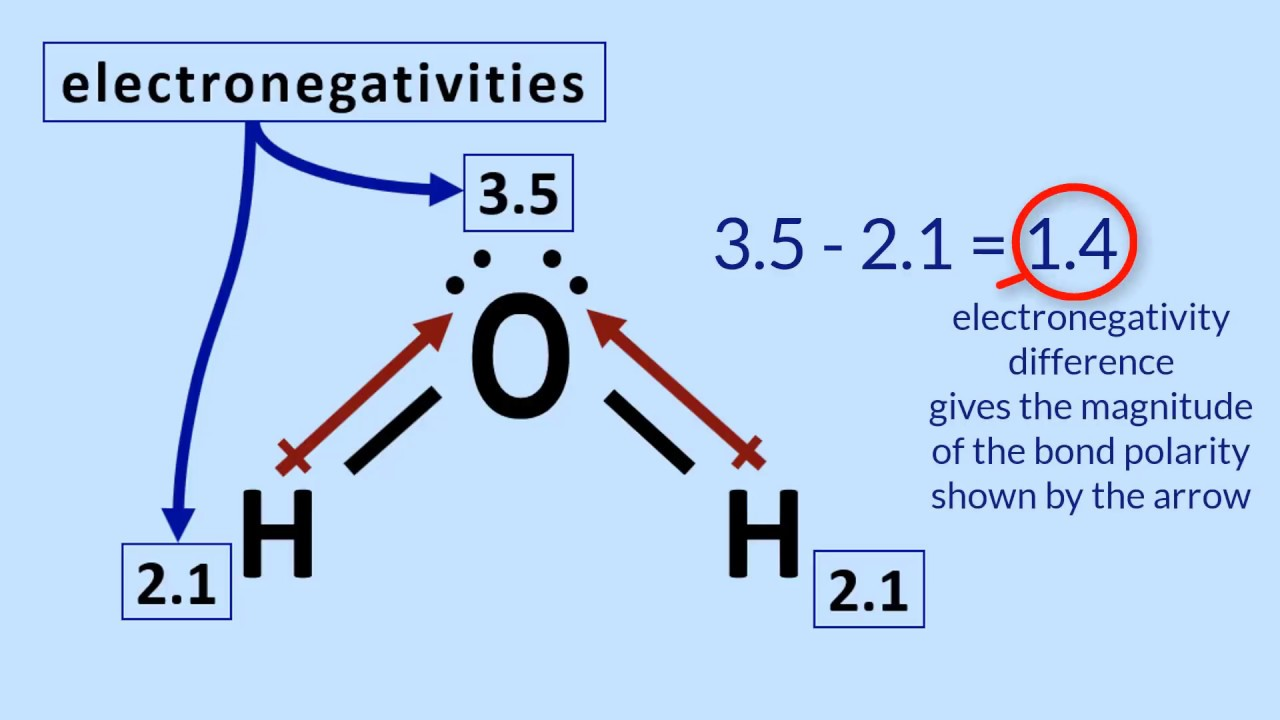
Apply the trend of atom electronegativity to calculate and/or determine the relative polarity of bonds and molecules.
Electronegativity increases across a period (left to right) and decreases down a group.
Find the difference in electronegativity between atoms:
0–0.4 → Nonpolar covalent
0.5–1.7 → Polar covalent
>1.7 → Ionic
To determine molecular polarity, check for bond dipoles and symmetry—if dipoles cancel, the molecule is nonpolar.
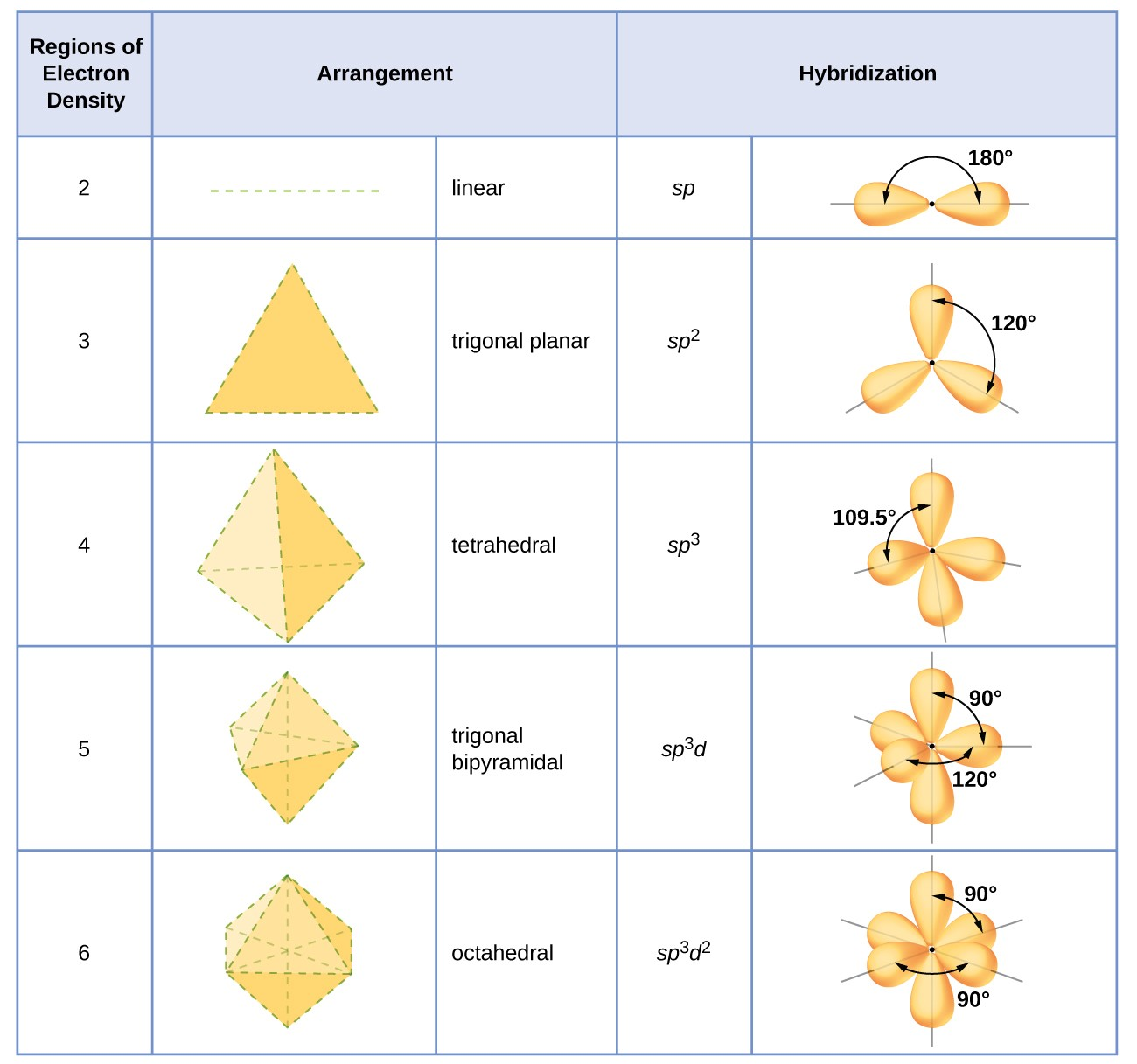
Explain hybridization, and recognize the type of hybridization in a given carbon center and the number of hybrid orbitals in a given structure.
Hybridization describes the mixing of atomic orbitals to form new hybrid orbitals for bonding:
sp³ (single bonds, tetrahedral, 109.5° bond angles)
sp² (one double bond, trigonal planar, 120° bond angles)
sp (one triple bond or two double bonds, linear, 180° bond angles)
Count sigma bonds + lone pairs to determine hybridization.
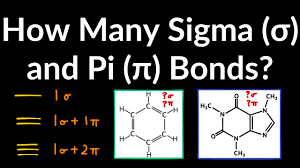
Identify the number of sigma and pi bonds in a structure.
Single bonds = 1 sigma (σ) bond
Double bonds = 1 sigma (σ) + 1 pi (π) bond
Triple bonds = 1 sigma (σ) + 2 pi (π) bonds
Count each bond in a structure to find total σ and π bonds.
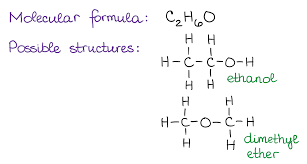
Determine the molecular formula of a given organic structure.
Count the number of C, H, O, N, etc. in the structure and write in CₓHᵧO𝓏... format.
Example: Ethanol C₂H₆O
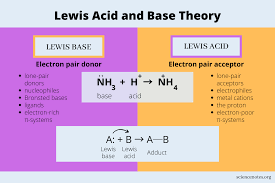
Identify and differentiate between Brønsted-Lowry acids and bases and Lewis acids and bases.
Brønsted-Lowry acid: Proton (H⁺) donor
Brønsted-Lowry base: Proton (H⁺) acceptor
Lewis acid: Electron pair acceptor
Lewis base: Electron pair donor
Example: NH₃ is a Brønsted base (accepts H⁺) and a Lewis base (donates electrons).
Brønsted: in terms of the proton, Lewis: in terms of the electron
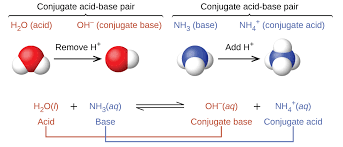
Classify the acid, base, conjugate acid, and conjugate base in a Brønsted-Lowry acid-base reaction.
Identify the acid (H⁺ donor) and the base (H⁺ acceptor).
The conjugate base is what remains after the acid donates H⁺.
The conjugate acid is what forms when the base gains H⁺.
Example:
HCl+H2O→Cl−+H3O+HCl + H₂O → Cl⁻ + H₃O⁺HCl+H2O→Cl−+H3O+Acid: HCl
Base: H₂O
Conjugate base: Cl⁻
Conjugate acid: H₃O⁺
Identify and/or rank relative acidity or basicity given the pKa.
Lower pKa = stronger acid (e.g., HCl, pKa ≈ -7).
Higher pKa = weaker acid (e.g., NH₃, pKa ≈ 38).
Compare pKa values to determine which acid is stronger.

Classify organic compounds according to their functional group.
Identify the key feature of the molecule (e.g., hydroxyl for alcohols, carbonyl for ketones).
Use structural formulas to match compounds to one of the 20 functional groups (e.g., alkene, ether, carboxylic acid).
Example: CH₃CH₂OH → Alcohol (hydroxyl group -OH).
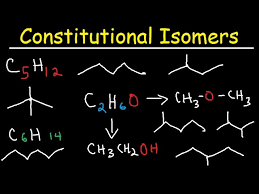
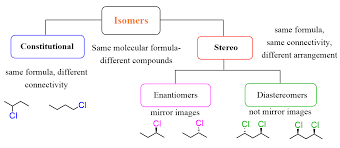
Identify constitutional (structural) isomers.
Constitutional isomers have the same molecular formula but different connectivity of atoms.
To identify:
Count the total number of C, H, O, etc. to ensure the formula matches.
Compare bonding patterns—different arrangements mean they are isomers.
Example: C₄H₁₀
n-butane: Straight-chain
isobutane: Branched
Draw the condensed and line structure for constitutional isomers of alkanes and cycloalkanes given their molecular formula and write the molecular formula of a compound given the structure.
Condensed structure: Write bonds without explicitly showing C-H bonds (e.g., CH₃CH₂CH₃ for propane).
Line structure: Use zigzag lines where each vertex represents a carbon, and hydrogen atoms are implied.
Given a structure, count all atoms to determine the molecular formula.
Name alkanes and cycloalkanes.
Find the longest chain → base name (meth-, eth-, prop-, etc.).
Number the chain so that the substituents get the lowest possible numbers.
Identify and name substituents (e.g., methyl, ethyl).
For cycloalkanes, use "cyclo-" before the base name.
Example: 2-methylpentane
Longest chain: pentane
Methyl group on C2
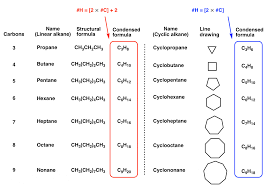
Classify carbons in a compound as primary, secondary, tertiary, or quaternary.
Primary (1°) carbon: Attached to one other carbon.
Secondary (2°) carbon: Attached to two carbons.
Tertiary (3°) carbon: Attached to three carbons.
Quaternary (4°) carbon: Attached to four carbons.
Example: (CH₃)₃CBr → Central carbon is tertiary (3°).
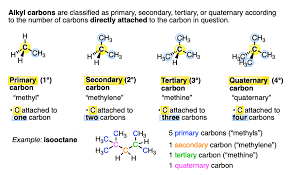
Describe properties of alkanes, such as boiling point and polarity.
Boiling point:
Increases with longer chains (more dispersion forces).
Decreases with more branching (less surface area for interactions).
Polarity:
Alkanes are nonpolar (only C-H bonds, no dipole moment).
Insoluble in water but soluble in nonpolar solvents.
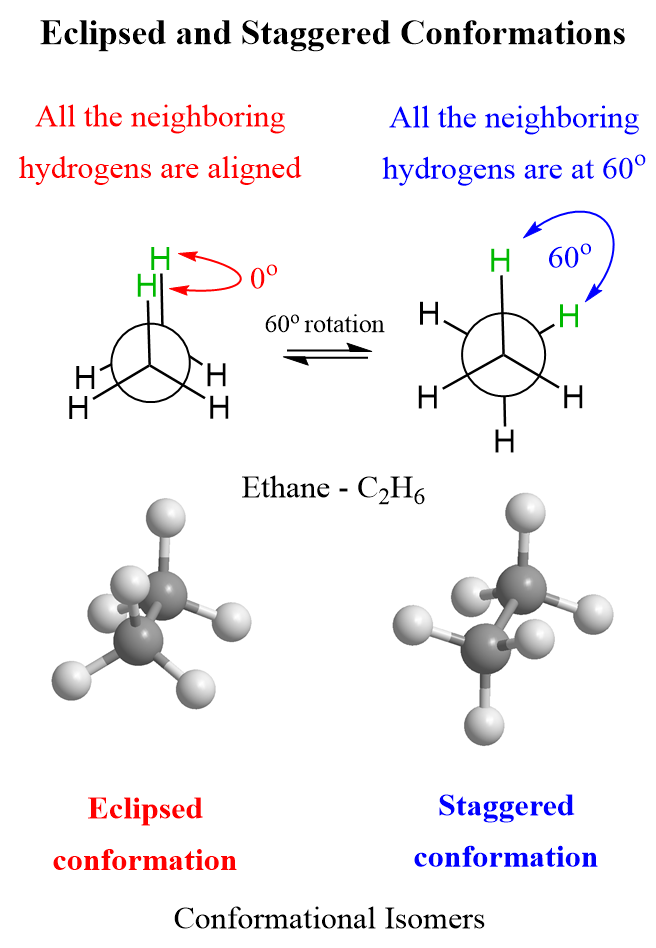
Draw conformational isomers of alkanes using Newman Projections.
Newman projections represent different rotations around single (sigma) bonds.
Looking down the C-C bond, draw the front carbon as a dot and the back carbon as a circle.
Draw three bonds from each carbon to represent attached groups.
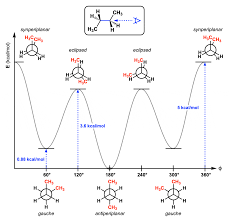
Identify staggered and eclipsed conformational isomers and describe their relative stability.
Staggered: Groups are as far apart as possible, making it more stable (lower energy).
Eclipsed: Groups overlap, creating torsional strain (less stable, higher energy).
Anti-staggered (largest groups opposite) is the most stable conformation.
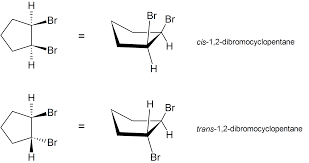
Draw and/or identify cis- and trans- isomers for disubstituted cycloalkanes.
Cis-isomer: Substituents are on the same side of the ring.
Trans-isomer: Substituents are on opposite sides of the ring.
Example: 1,2-dimethylcyclohexane
Cis: Both methyl groups point up or down.
Trans: One methyl points up, the other down.
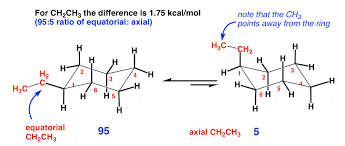
Identify whether substituents are in the axial or equatorial position in cyclohexane and determine the most stable conformational isomer for mono- and di-substituted rings.
Axial: Substituents point straight up/down.
Equatorial: Substituents point slightly outward, making them more stable.
Most stable conformation: Larger groups prefer the equatorial position to avoid steric strain.
Example: tert-butylcyclohexane → tert-butyl group is equatorial in the most stable form.
Identify chirality centers at tetrahedral carbons.
A chirality center (chiral carbon) is a tetrahedral carbon (sp³ hybridized) bonded to four different groups.
To identify:
Locate all sp³ hybridized carbons.
Check if each carbon is bonded to four distinct groups (atoms, chains, or functional groups).
Example: 2-butanol (CH₃CHOHCH₂CH₃) → The second carbon is a chirality center since it is bonded to H, OH, CH₃, and CH₂CH₃.
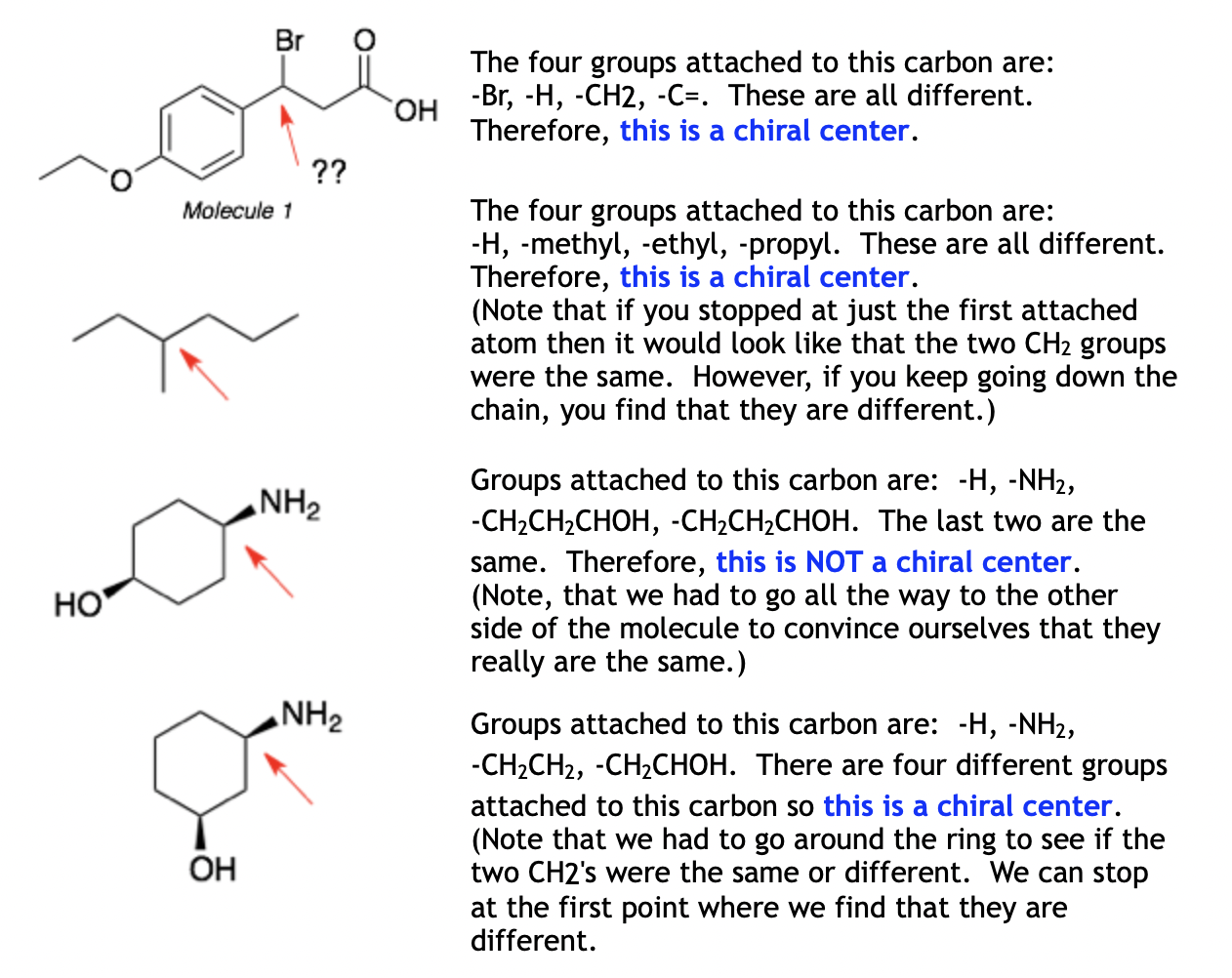
Determine the number of chiral centers present in a molecule.
Count all tetrahedral carbons with four different groups.
Double bonds or symmetrical carbons do not count.
If a molecule has n chiral centers, it can have up to 2ⁿ stereoisomers.
Example: Glucose (C₆H₁₂O₆) has four chiral centers, leading to 16 possible stereoisomers.
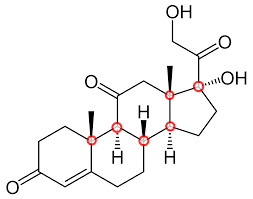
Recognize whether a molecule is chiral or achiral.
Chiral molecules: Have at least one chiral center and no plane of symmetry.
Achiral molecules: Either lack chirality centers or have a plane of symmetry, meaning they are superimposable on their mirror image.
Example: 2-chlorobutane is chiral (one chiral center), but 2,2-dichlorobutane is achiral (symmetry).
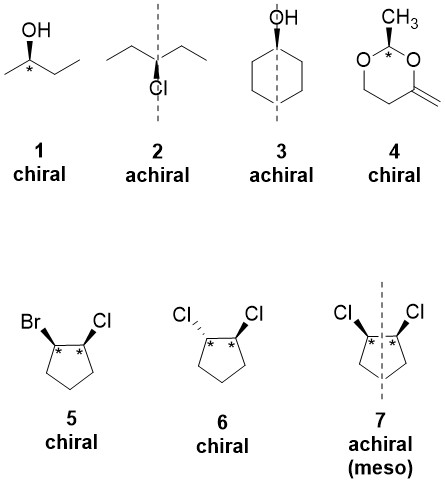
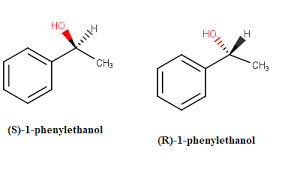
Apply Cahn-Ingold-Prelog sequence rules to rank and determine the R/S configuration of chirality centers.
Step 1: Assign priorities (1 → highest, 4 → lowest) based on atomic number of directly attached atoms.
Step 2: If there’s a tie, move to the next atom in the chain.
Step 3: Orient the molecule so that priority 4 is in the back (dashed line).
Step 4: Draw a curved arrow from priority 1 → 2 → 3.
Clockwise → R configuration.
Counterclockwise → S configuration.
Identify the isomeric relationship of a pair of compounds as enantiomers, diastereomers, the same or constitutional isomers.
Enantiomers: Non-superimposable mirror images (all chiral centers have opposite R/S).
Diastereomers: Not mirror images, differ at some but not all chiral centers.
Same compound: Identical R/S configuration and connectivity.
Constitutional isomers: Same molecular formula, but different connectivity.
Example: (R)-2-butanol vs. (S)-2-butanol → Enantiomers.
Draw a molecule with the correct stereochemistry.
Use wedges and dashes:
Solid wedge (▲) → Bond coming out of the plane.
Dashed wedge (▬) → Bond going behind the plane.
Follow R/S configuration rules when placing groups.
For Fischer projections, keep the horizontal bonds coming out and vertical bonds going back.
Example: Drawing (R)-2-butanol, ensure OH is in the correct position based on Cahn-Ingold-Prelog priority rules.
Name alkenes, cycloalkenes and alkynes using IUPAC nomenclature.
What: Systematically naming unsaturated hydrocarbons by identifying the carbon chain and location of multiple bonds.
How:
Identify the longest carbon chain that includes the double (alkene) or triple bond (alkyne).
Number the chain so that the multiple bond gets the lowest possible number.
Give priority alphabetically.
Use suffixes: “-ene” for alkenes and cycloalkenes, “-yne” for alkynes; for cyclic compounds, add the prefix “cyclo.”
Add -a to the numbered chain (ex: hept_a_) when followed by a consonant
Example:
Alkene: 2-butene (CH₃CH=CHCH₃)
Cycloalkene: Cyclohexene
Alkyne: 2-butyne (CH₃C≡CCH₃)
Identify and draw cis-/trans- and E/Z isomers of alkenes.
What: Recognizing stereoisomers based on the spatial arrangement of substituents around the double bond.
How:
Cis/Trans: Directly compare positions of identical substituents on each carbon.
E/Z: Apply the Cahn-Ingold-Prelog priority rules to assign priorities to substituents; if high-priority groups are on the same side, it’s Z (“zusammen”), and if opposite, it’s E (“entgegen”).
Example:
Cis-2-butene: Both CH₃ groups are on the same side of the double bond.
Trans-2-butene: CH₃ groups are on opposite sides.
For dissimilar substituents, assign E or Z based on the priorities determined.
Describe electrophiles and nucleophiles in chemical reactions.
What: Electrophiles are electron-poor species that seek electrons, while nucleophiles are electron-rich species that donate electrons.
How:
Identify electrophiles by their partial positive charge or electron deficiency (often featuring an empty orbital).
Recognize nucleophiles by their negative charge or presence of lone pairs available for donation.
Example:
In the addition of HBr to an alkene, the H⁺ acts as the electrophile (seeking electrons), and the alkene’s π electrons serve as the nucleophile.
Classify the types of organic reactions, such as additions, eliminations, substitutions, and rearrangements
What: Categorizing reactions based on changes in bonding: additions, eliminations, substitutions, and rearrangements.
How:
Addition: Atoms or groups are added to a molecule (e.g., hydrogenation).
Elimination: Atoms or groups are removed from a molecule (e.g., dehydrohalogenation).
Substitution: One atom or group is replaced by another (e.g., SN1/SN2 reactions).
Rearrangement: Structural reorganization within the molecule (e.g., carbocation shifts).
Example:
Addition: Hydrogenation of ethene (adding H₂).
Elimination: Formation of propene from isopropyl bromide via dehydrohalogenation.
Substitution: SN2 reaction of methyl bromide with hydroxide ion (OH⁻).
Rearrangement: Hydride shift during carbocation rearrangement in a reaction intermediate.
Determine whether a reaction proceeds by a homolytic or heterolytic mechanism.
What:
Homolytic cleavage: Bond breaks evenly, with each atom retaining one electron, forming radicals.
Heterolytic cleavage: Bond breaks unevenly, with one atom taking both electrons, forming ions.
How:
Examine the reaction conditions: radical initiators (light, heat, peroxides) often indicate homolytic mechanisms; polar solvents and strong acids/bases favor heterolytic pathways.
Example:
Homolytic: Chlorination of methane under UV light, forming methyl radicals.
Heterolytic: Dissociation of HCl in water, yielding H⁺ and Cl⁻ ions.
Predict products and draw the mechanism for the hydrohalogenation of alkenes.
What:
Addition of a hydrogen halide (HX) to an alkene that follows Markovnikov’s rule.
How:
Step 1: The alkene’s π electrons attack H⁺ from HX, forming the most stable carbocation intermediate.
Step 2: The halide ion (X⁻) then attacks the carbocation to yield the haloalkane.
Example:
Propene + HBr:
Protonation at the less substituted carbon forms a secondary carbocation.
Bromide ion attacks to produce 2-bromopropane.
Mechanism diagram:
Propene → (H⁺ addition) → Carbocation intermediate → (Br⁻ attack) → 2-bromopropane.
Identify reactants, products, intermediates, transition states, activation energy (ΔG‡), and Gibbs Free Energy (ΔG°) on a reaction diagram.
What:
A reaction coordinate diagram shows the energy profile of a reaction, marking reactants, products, intermediates, transition states, and energy changes.
How:
Plot energy (vertical axis) versus reaction progress (horizontal axis).
Mark the highest point as the transition state (ΔG‡, the activation energy) and note the overall energy difference between reactants and products (ΔG°).
Example:
For a hydrohalogenation reaction:
Reactants: Alkene + HX
Intermediate: Carbocation formed after protonation
Transition State: The energy peak during carbocation formation
Product: Haloalkane; the diagram shows an exothermic overall ΔG° if product energy is lower than that of the reactants.
Apply Markonikov’s rule to the reactions of alkenes to predict the correct regioisomer.
What:
Markovnikov’s rule predicts that in the addition of HX to an unsymmetrical alkene, the hydrogen atom bonds to the carbon with more hydrogen atoms, while the halide attaches to the more substituted carbon.
How:
Identify the alkene’s substituents; predict which pathway leads to the more stable (more substituted) carbocation.
Example:
1-Butene + HBr:
Hydrogen adds to the terminal CH₂, forming a secondary carbocation that is more stable, leading to the formation of 2-bromobutane rather than 1-bromobutane.
Rank the relative stability of carbocations.
What:
Carbocations are classified by their substitution level; tertiary (3°) are most stable, followed by secondary (2°), then primary (1°).
How:
Evaluate based on hyperconjugation and inductive effects from alkyl groups.
Example:
Tertiary Carbocation: (CH₃)₃C⁺ is more stable than
Secondary Carbocation: CH₃CH(CH₃)⁺, which in turn is more stable than
Primary Carbocation: CH₃CH₂⁺.
Predict the product and explain the following alkene addition reactions.
a. Hydrohalogenation (mechanism) - addition of HX
b. Acid catalyzed hydration (mechanism) - addition of water
c. Halogenation - addition of Br2 or Cl2
d. Hydrogenation - addition of H2
a. Hydrohalogenation (Addition of HX)
What:
Addition of HX to an alkene following Markovnikov’s rule.
How:
Protonate the double bond to form a carbocation; then, the halide ion attacks the carbocation.
Example:
Ethene + HBr: Leads to bromoethane.
b. Acid-Catalyzed Hydration (Addition of Water)
What:
Addition of water to an alkene in the presence of an acid catalyst to form an alcohol.
How:
Protonation of the alkene forms a carbocation; water attacks the carbocation; a deprotonation step gives the alcohol.
Example:
Propene + H₂O (acid catalyst): Forms 2-propanol.
c. Halogenation (Addition of Br₂ or Cl₂)
What:
Addition of a halogen molecule to an alkene forming a dihalide.
How:
Formation of a cyclic halonium ion intermediate; subsequent attack by a halide ion opens the ring.
Example:
Cyclohexene + Br₂: Yields trans-1,2-dibromocyclohexane.
d. Hydrogenation (Addition of H₂)
What:
Addition of hydrogen to an alkene, typically catalyzed by a metal such as Pd or Pt, converting the double bond to a single bond.
How:
Both the alkene and H₂ adsorb onto the catalyst surface; hydrogen atoms add to the double bond sequentially.
Example:
Ethene + H₂ (with Pd catalyst): Produces ethane.
Identify conjugated dienes and describe their properties.
What:
Conjugated dienes feature alternating double and single bonds (e.g., C=C–C=C).
Properties:
They exhibit resonance stabilization and lower reactivity toward electrophilic addition than isolated double bonds.
Often show characteristic UV absorption due to extended π-electron delocalization.
Example:
1,3-Butadiene (CH₂=CH–CH=CH₂).
Predict the product and explain the following alkyne addition reactions.
a. Hydrohalogenation- addition of HX
b. Acid catalyzed hydration - addition of water
c. Halogenation- addition of Br2 or Cl2
d. Hydrogenation - addition of H2
a. Hydrohalogenation – Addition of HX
What:
HX adds to an alkyne in a stepwise fashion, following Markovnikov’s rule.
Mechanism:
Step 1: Protonation of the alkyne forms a vinyl cation intermediate.
Step 2: Halide ion (X⁻) attacks the carbocation; with excess HX, a second addition can yield a geminal dihalide.
Example:
1-Butyne + HBr: Initial formation of a bromoalkene, which may further add HBr to give 2,2-dibromobutane.
b. Acid-Catalyzed Hydration – Addition of Water
What:
Water adds to an alkyne in the presence of acid, forming an enol that tautomerizes to a carbonyl compound.
Mechanism:
Step 1: Protonation forms a vinyl cation.
Step 2: Water attacks the carbocation, followed by deprotonation.
Step 3: The resulting enol rapidly tautomerizes to a ketone (or aldehyde).
Example:
2-Butyne + H₂O/H₂SO₄ → 2-Butanone.
c. Halogenation – Addition of Br₂ or Cl₂
What:
Halogen addition to an alkyne can occur stepwise.
Mechanism:
Step 1: Formation of a cyclic halonium ion intermediate.
Step 2: Nucleophilic attack by a halide ion leads to a dihalogenated alkene; with excess halogen, a tetrahalogenated product may form.
Example:
2-Butyne + Br₂ (1 equiv.) → 1,2-Dibromo-2-butene; excess Br₂ can yield a tetrahalide.
d. Hydrogenation – Addition of H₂
What:
Catalytic hydrogenation converts alkynes to alkenes or fully to alkanes.
Mechanism:
On a metal catalyst surface, hydrogen atoms add to the triple bond.
Use of a poisoned catalyst (e.g., Lindlar’s catalyst) stops at the alkene stage, while a non-poisoned catalyst leads to complete hydrogenation.
Example:
2-Butyne + H₂ with Lindlar’s catalyst → cis-2-Butene; complete hydrogenation yields butane.
Name alkyl halide molecules using IUPAC nomenclature.
What:
Identify the longest carbon chain containing the halogen; number the chain to give the halogen the lowest possible number.
How:
Use the suffix “-haloalkane” or the halogen prefix (e.g., chloro-, bromo-) followed by the parent alkane name.
Example:
CH₃CH₂CH₂Cl is named 1-chloropropane.
Prepare alkyl halide compounds from radical halogenation and alkene addition reactions.
Radical Halogenation:
What:
Direct substitution of a hydrogen atom in an alkane by a halogen atom under UV light.
Mechanism:
Initiation (formation of radicals), propagation (abstraction of hydrogen and formation of alkyl radical, which reacts with X₂), and termination.
Example:
Methane + Cl₂ (UV light) → Chloromethane.
Alkene Addition Reaction:
What:
Addition of HX to an alkene follows Markovnikov’s rule.
Example:
Ethene + HBr → Bromoethane.
Prepare alkyl halides by reaction of an alcohol with thionyl chloride or PBr3.
What:
Convert an alcohol into its corresponding alkyl halide by substituting the –OH group with a halogen.
How:
The alcohol reacts with thionyl chloride (SOCl₂) to form an alkyl chloride, or with phosphorus tribromide (PBr₃) to form an alkyl bromide.
Example:
Ethanol + SOCl₂ → Chloroethane; or Ethanol + PBr₃ → Bromoethane.
Identify alcohols as primary, secondary, or tertiary.
What:
Primary: –OH attached to a carbon bonded to one other carbon.
Secondary: –OH attached to a carbon bonded to two other carbons.
Tertiary: –OH attached to a carbon bonded to three other carbons.
How:
Examine the carbon bearing the –OH group and count its carbon neighbors.
Example:
Methanol (CH₃OH): Primary
Isopropanol (CH₃CHOHCH₃): Secondary
tert-Butanol ((CH₃)₃COH): Tertiary
Differentiate between elimination and substitution reactions.
What:
Substitution: One functional group is replaced by another.
Elimination: Atoms or groups are removed, forming double or triple bonds.
How:
Look for replacement (substitution) versus removal with formation of unsaturation (elimination).
Example:
Substitution: SN2 reaction of CH₃Br with OH⁻ gives CH₃OH.
Elimination: Dehydrohalogenation of CH₃CH₂Br with a strong base produces ethene.
Identify the nucleophile, the substrate, and the leaving group for substitution and elimination reactions
Nucleophile: Electron-rich species that attacks an electrophilic center.
Substrate: Molecule containing the leaving group, undergoing reaction.
Leaving Group: Atom or group that departs with its bonding electrons.
Example:
In the SN2 reaction of CH₃Br with OH⁻:
Nucleophile: OH⁻
Substrate: CH₃Br
Leaving Group: Br⁻
Prepare alkyl halides from the reaction of alcohols with HX.
What:
Replace the –OH group in an alcohol with a halide from HX.
How:
The reaction proceeds via SN1 (tertiary) or SN2 (primary/secondary) pathways.
Example:
Ethanol + HCl → Chloroethane
Describe biological pathways that utilize SN1 and SN2 mechanisms.
SN1: Two-step reaction (carbocation forms first).
Favored by tertiary carbons, weak nucleophiles, and polar protic solvents.
Example: Steroid biosynthesis (carbocation rearrangements).
SN2: One-step reaction (nucleophile attacks, leaving group exits simultaneously).
Favored by primary carbons, strong nucleophiles, and polar aprotic solvents.
Example: DNA methylation via SAM, glycosyl transfer in metabolism.
Key Differences:
SN1: Carbocation, racemization, slower first step.
SN2: Backside attack, inversion of configuration, no intermediates.
Identify the major organic product and determine which reaction pathway is favored for E1, E2, SN1and SN2 reactions given a set of reaction conditions.
What:
Reaction conditions (solvent, temperature, nucleophile/base strength, substrate structure) determine whether E1, E2, SN1, or SN2 dominates.
How:
E1/SN1: Favored by tertiary substrates, weak nucleophiles, polar protic solvents, and moderate temperatures.
E2/SN2: Favored by strong bases/nucleophiles, primary or secondary substrates, and polar aprotic solvents.
Example:
2-Bromopropane with a strong base (e.g., KOH at high temperature): Likely undergoes E2 elimination, while a weak nucleophile under milder conditions may favor SN1 substitution.
Use Zaitsev’s rule to predict major organic products of elimination reactions.
What:
Zaitsev’s rule states that the more substituted (and more stable) alkene is usually the major product.
How:
Compare possible alkene products; the one with the greater number of alkyl substituents on the double bond is favored.
Example:
2-Bromobutane elimination: Predominantly forms 2-butene (more substituted) over 1-butene.
Predict relative rates of reactivity of substrates with alkyl halides from E1, E2, SN1 and SN2
Predicting Relative Rates of Reactivity in E1, E2, SN1, and SN2
What:
Reactivity depends on substrate structure, nucleophile/base strength, leaving group ability, and solvent.
How:
SN1/E1: Tertiary > secondary > primary (due to carbocation stability).
SN2: Primary > secondary; tertiary substrates are typically unreactive due to steric hindrance.
E2: Requires a strong base and available β-hydrogens; rate increases with substrate substitution (if sterics don’t overly hinder the base).
Example:
Tertiary alkyl halide: Fast SN1/E1 reaction due to stable carbocation.
Primary alkyl halide: Fast SN2 reaction due to minimal steric hindrance, but slow SN1 due to unstable carbocation formation.
Identify aromatic compounds.
Must follow Hückel's Rule: planar, cyclic, fully conjugated π system with (4n + 2) π electrons (n = integer).
Benzene (6 π electrons) is the classic example.
Commonly seen: toulene, benzoic acid, aniline, acetophenone, styrene, benzoic acid, phenol, benzaldehyde
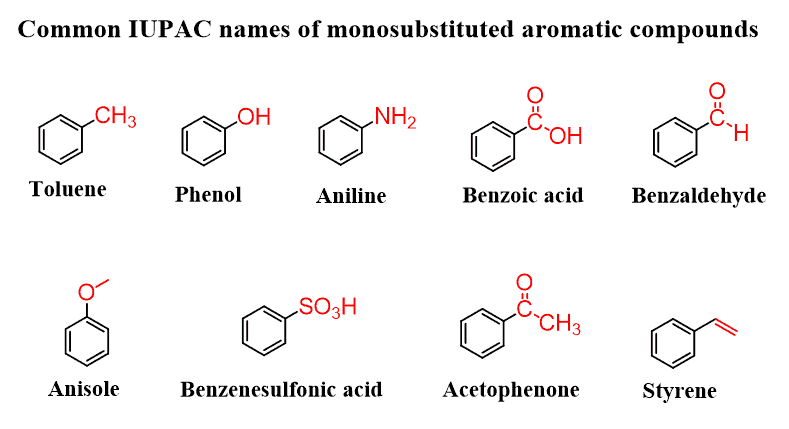
Name aromatic compounds using common names and IUPAC nomenclature.
Common names: toluene (methylbenzene), aniline (aminobenzene), phenol (hydroxybenzene).
IUPAC: treat benzene as the parent ring; name substituents with positions (e.g., 1-bromo-3-nitrobenzene).
Use ortho (o-), meta (m-), and para (p-) for disubstituted benzenes (1,2; 1,3; 1,4 positions)
Predict the product and identify the active electrophile for the following electrophilic aromatic substitution reactions.
a. Bromination/Chlorination
b. Nitration
c. Sulfonation
d. Friedel-Crafts Alkylation
e. Friedel-Crafts Acylation
a. Bromination/Chlorination
Reagents: Br₂ or Cl₂ with FeBr₃ or AlCl₃.
Electrophile: Br⁺ / Cl⁺.
b. Nitration
Reagents: HNO₃ + H₂SO₄.
Electrophile: NO₂⁺ (nitronium ion).
c. Sulfonation
Reagents: SO₃ + H₂SO₄ or fuming H₂SO₄.
Electrophile: SO₃H⁺.
d. Friedel-Crafts Alkylation
Reagents: R–Cl + AlCl₃.
Electrophile: carbocation (R⁺) or rearranged carbocation.
e. Friedel-Crafts Acylation
Reagents: RCOCl + AlCl₃.
Electrophile: acylium ion (R–C⁺=O).
Predict the major organic product for EAS reactions of monosubstituted benzene derivatives by identifying if the substituent is o/p- or m-directing.
o/p-directors (activators): –OH, –OCH₃, –NH₂, –CH₃.
m-directors (deactivators): –NO₂, –CF₃, –SO₃H, –CN.
Directing group determines where new substituent adds on the ring.
Rank relative reactivity of substituted benzene derivatives by identifying if the substituent on the benzene ring is an activator or deactivator.
Activators: donate e⁻ via resonance/induction → speed up EAS.
Example: –OH > –NH₂ > –CH₃.
Deactivators: withdraw e⁻ → slow EAS.
Example: –NO₂ > –COOH > –CF₃.
Halogens are weird: deactivating but o/p-directing.
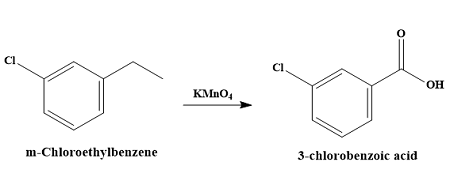
Predict the product for oxidations of alkyl benzenes with potassium permanganate or sodium dichromate
Reagents: KMnO₄, Na₂Cr₂O₇, or similar.
Converts any benzylic –CH₃, –CH₂R group to –COOH, if there is at least one benzylic hydrogen.
Benzene ring remains untouched.
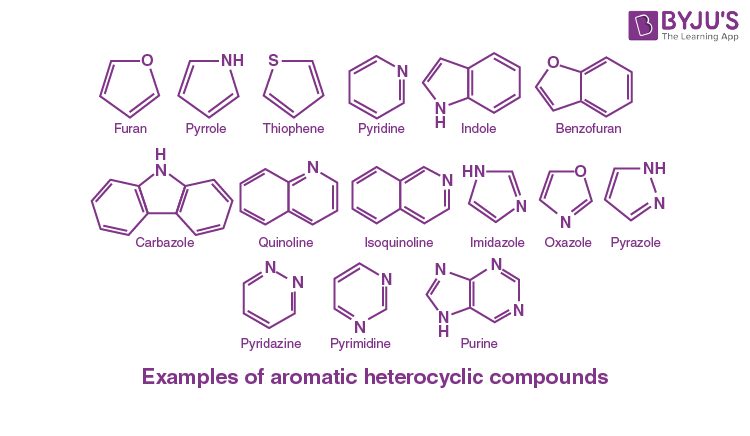
Identify heterocyclic aromatic compounds
Cyclic, aromatic rings containing atoms other than carbon (e.g., N, O, S).
Must follow Hückel’s Rule for aromaticity.
Examples: pyridine, furan, thiophene, pyrrole.
Identify a short multistep synthesis route for an organic target.
Work backward using retrosynthesis.
Identify:
Key bonds to form.
Functional group transformations.
Order of reactions (directing effects matter!).
Use EAS + oxidations + Friedel-Crafts combos.
Identify the following functional groups: alcohols, thiols, ethers, and sulfides.
Alcohol: –OH (hydroxyl group on sp³ C).
Thiols: –SH (like alcohols but with sulfur).
Ethers: R–O–R′.
Sulfides: R–S–R′ (sulfur analog of ether).
Name alcohols, thiols, ethers and sulfides with IUPAC nomenclature.
Alcohols: replace “-e” with “-ol” (e.g., propan-2-ol).
Thiols: add “-thiol” to parent name (e.g., ethanethiol).
Ethers/Sulfides: name smaller group as alkoxy/alkylthio substituent (e.g., methoxyethane or methylthioethane).
Classify an alcohol as methyl, primary, secondary, or tertiary.
Methyl: only one carbon (CH₃OH).
Primary (1°): –OH on C bonded to 1 other C.
Secondary (2°): –OH on C bonded to 2 other Cs.
Tertiary (3°): –OH on C bonded to 3 other Cs.
Describe the physical properties of alcohols.
High boiling point due to hydrogen bonding.
More soluble in water if small.
Longer hydrocarbon chains reduce polarity.
Describe spectroscopy techniques to identify and characterize alcohols.
IR: broad O–H stretch around 3200–3600 cm⁻¹.
¹H NMR: O–H proton shows up as a broad singlet, usually 1–5 ppm.
¹³C NMR: alcohol carbon near 60 ppm.
Synthesize alcohols using the following reactions:
a. Acid-catalyzed hydration
b. Substitution reaction
c. Reduction of carbonyl compound
d. Grignard reaction
a. Acid-Catalyzed Hydration
Alkene + H₂O + H₂SO₄ → Markovnikov alcohol.
b. Substitution Reaction
SN1/SN2: alkyl halide + H₂O or OH⁻ → alcohol.
c. Reduction of Carbonyl
Aldehyde/ketone + NaBH₄ or LiAlH₄ → alcohol.
d. Grignard Reaction
RMgX + aldehyde/ketone → alcohol (after H₃O⁺ workup).
Formaldehyde → primary alcohol
Aldehyde → secondary
Ketone → tertiary
Predict the product, starting material, or reagents needed for the following reactions of alcohols:
a. Dehydration
b. Oxidation with a mild oxidant
c. Oxidation with a strong oxidant
d. Williamson ether synthesis
a. Dehydration
Alcohol + H₂SO₄, heat → alkene (E1 for 2°/3°, E2 for 1°).
b. Oxidation (Mild: PCC)
1° alcohol → aldehyde
2° alcohol → ketone
3° alcohol → no reaction
c. Oxidation (Strong: KMnO₄ or CrO₃)
1° alcohol → carboxylic acid
2° alcohol → ketone
d. Williamson Ether Synthesis
RO⁻ + R′–X (1° alkyl halide) → R–O–R′ (ether).
Predict the product, starting material or reactant for the epoxidation of an alkene with a peroxy acid.
Alkene + mCPBA (or other peroxy acid) → epoxide (triangle ring with O).
Draw the epoxide ring-opening product with HX or H3O+
With HX: opens to trans product, X on more substituted C (acidic conditions).
With H₃O⁺: OH groups add anti across ring, more stable C gets attacked.
Synthesize thiols by the reaction of alkyl halides with NaSH in an SN2 reaction.
R–X + NaSH → R–SH
Classic SN2: inversion of configuration if chiral.
Synthesize sulfides in an analogous reaction to the Williamson ether synthesis
R–X + R′–S⁻ → R–S–R′
Use thiolate (R′–S⁻) as nucleophile in SN2.
Identify aldehyde and ketone functional groups
Aldehyde: Carbonyl (C=O) bonded to at least one H (–CHO).
Ketone: Carbonyl bonded to two carbon groups (R–CO–R′)
Name aldehydes and ketones using IUPAC nomenclature.
Aldehyde: Replace “-e” with “-al” (e.g., ethanal).
Ketone: Replace “-e” with “-one”, number to give carbonyl lowest possible number (e.g., pentan-2-one).
Describe spectroscopy techniques to identify and characterize aldehydes and ketones
IR: Strong C=O stretch around 1700 cm⁻¹.
Aldehydes also have C–H stretch ~2720 cm⁻¹.
¹H NMR: Aldehyde H shows ~9–10 ppm.
¹³C NMR: Carbonyl C shows up ~190–210 ppm.
Synthesize a ketone by the following reactions:
a. Hydration of alkyne
b. Friedel-Craft alkylation
c. Oxidation of an alcohol
a. Hydration of Alkyne
Terminal/internal alkyne + H₂O, H₂SO₄, HgSO₄ → enol → ketone (via tautomerization).
b. Friedel-Crafts Acylation
Benzene + RCOCl + AlCl₃ → aryl ketone.
c. Oxidation of Secondary Alcohol
2° alcohol + PCC, Na₂Cr₂O₇, or KMnO₄ → ketone.
Synthesize an aldehyde by the following reactions:
a. Oxidation of an alcohol
a. Oxidation of Primary Alcohol
1° alcohol + PCC → aldehyde.
Strong oxidants (e.g., KMnO₄) take it all the way to a carboxylic acid.
Identify the electrophile and nucleophile in nucleophilic addition reactions of aldehydes and ketones
Electrophile: Carbon of the carbonyl (C=O).
Nucleophile: Anything that attacks the carbonyl (e.g., H⁻, R⁻, NH₃, H₂O).
Identify primary, secondary, and tertiary amines.
Primary (1°): R–NH₂
Secondary (2°): R₂NH
Tertiary (3°): R₃N
Identify acetal, hemiacetal, imine, and hydrate functional groups.
Acetal: Carbon bonded to 2 OR groups and 1 H or R (from aldehyde/ketone).
Hemiacetal: One OH and one OR group.
Imine: C=N–R (from reaction with 1° amine).
Hydrate: Carbon with two –OH groups (gem-diol).
Predict the product, starting material and/or reagents for the following reactions of aldehydes and ketones.
a. Reduction
b. Grignard
c. Hydrate formation
d. Acetal formation
e. Imine formation
a. Reduction
Aldehyde → 1° alcohol
Ketone → 2° alcohol
Reagents: NaBH₄, LiAlH₄, or catalytic H₂/Pd.
b. Grignard
RMgX + carbonyl → alcohol after H₃O⁺.
Aldehyde → 2° alcohol
Ketone → 3° alcohol
c. Hydrate Formation
Aldehyde/ketone + H₂O ⇌ hydrate (gem-diol).
Reversible, favored for small/very reactive carbonyls.
d. Acetal Formation
Carbonyl + 2 ROH + acid catalyst ⇌ acetal.
Use excess alcohol to drive forward.
Acetals are stable protecting groups.
e. Imine Formation
Carbonyl + 1° amine + acid catalyst ⇌ imine (C=N).
Water is a byproduct.
Identify carboxylic acids, acid halides, acid anhydrides, amides, esters, and nitriles, and be able to name carboxylic acids using current IUPAC nomenclature.
Carboxylic Acids: R-COOH; name ends in -oic acid (e.g., ethanoic acid).
Acid Halides: R-COX (X = Cl, Br); name as alkanoyl halide (e.g., ethanoyl chloride).
Anhydrides: R-CO-O-COR; name as alkanoic anhydride (e.g., acetic anhydride).
Esters: R-COOR'; name as alkyl alkanoate (e.g., methyl ethanoate).
Amides: R-CONH₂, R-CONHR’, R-CONR₂; name as alkanamide.
Nitriles: R-C≡N; name as alkanenitrile (e.g., ethanenitrile).
Recognize the general acidity of carboxylic acids in acid-base reactions.
Carboxylic acids are weak acids (pKa ≈ 4–5).
Resonance stabilizes the carboxylate anion, making them more acidic than alcohols.
Determine the product, starting material, and/or reagents for the following reactions to synthesize carboxylic acids:
a. Alkyl benzene oxidation
b. Alcohol and aldehyde oxidation
c. Hydrolysis of carboxylic acid derivatives
a. Alkyl Benzene Oxidation: Use KMnO₄ or H₂CrO₄ to oxidize benzylic carbon to COOH.
b. Alcohol & Aldehyde Oxidation: 1° alcohol or aldehyde + Jones reagent or KMnO₄ → COOH.
c. Hydrolysis of Derivatives: Acid/base hydrolysis of esters, nitriles, amides → COOH.
Determine the product, starting material, and/or reagents for reactions of carboxylic acids to produce acyl chlorides or esters.
To Acyl Chloride: Use SOCl₂, PCl₃, or PCl₅.
To Ester: Use alcohol + acid catalyst (Fischer esterification).
Draw the product, starting material, and/or reagents for the following nucleophilic acyl substitution reactions:
a. Acid Halides conversion to:
ii. Ester
iii. Amide
iv. Alcohol
b. Acid anhydride conversion to:
i. Ester
ii. Amide
iii. Alcohol
c. Ester conversion to:
i. Amide
ii. Alcohol
d. Amide conversion to
i. Amine
a. Acid Halides →
Ester: Add alcohol.
Amide: Add ammonia or amine.
Alcohol: Add water (hydrolysis).
b. Acid Anhydrides →
Ester: Add alcohol.
Amide: Add ammonia or amine.
Alcohol: Add water (hydrolysis).
c. Esters →
Amide: Add ammonia or amine (requires heating).
Alcohol: Add water + acid/base (hydrolysis).
d. Amides →
Amine: Use LiAlH₄ to reduce to amine.
Describe various applications that use carboxylic acid derivative chemistry.
Used in pharmaceuticals (e.g., aspirin = ester).
Important for polymer synthesis (e.g., nylon from diamides).
Widely used in fragrances, soaps, and plasticizers.
Identify a suitable route in a short multistep synthesis of an organic target.
Identify target functional group; work backward (retrosynthesis).
Use known conversions (e.g., alcohol → ester → carboxylic acid).
Minimize steps and choose conditions compatible with all functional groups.
Identify and name amines.
Amines: Nitrogen bonded to alkyl/aryl groups (R-NH₂, R₂NH, R₃N).
Naming:
IUPAC: Identify longest chain, use suffix “-amine” (e.g., ethanamine).
Common: Name alkyl groups alphabetically + “amine” (e.g., dimethylamine)
Describe properties of amines.
Boiling Point: Higher than alkanes, lower than alcohols (H-bonding in 1° & 2°).
Solubility: Small amines are water-soluble due to H-bonding.
Odor: Often fishy or unpleasant smell.
Rank the basicity of different amines.
Rank: 2° > 1° > 3° (in aqueous solution, due to solvation).
Aromatic Amines (e.g., aniline): Less basic due to resonance with benzene ring.
Electron-donating groups ↑ basicity; withdrawing groups ↓ basicity.
Describe spectroscopy techniques to identify and characterize amines.
IR:
1° amine → 2 N-H stretches (~3300 & 3400 cm⁻¹).
2° amine → 1 N-H stretch.
NMR: N-H proton = broad peak (~1-5 ppm); adjacent C-H shifts slightly downfield.
Mass Spec: Characteristic fragmentation near the nitrogen.
Predict the product, starting material and/or reagents for the following reactions to produce amines.
a. Reduction of nitriles
b. Reduction of amides
c. Reduction of azides
d. Reductive amination
e. Reduction of nitrobenzene
a. Reduction of Nitriles
Reagents: LiAlH₄ or H₂/Pd.
Product: 1° amine (R-CN → R-CH₂NH₂).
b. Reduction of Amides
Reagents: LiAlH₄.
Product: 1°, 2°, or 3° amines depending on R-groups.
c. Reduction of Azides
Reagents: H₂/Pd or LiAlH₄.
Product: 1° amine (RN₃ → RNH₂).
d. Reductive Amination
Reagents: Aldehyde/ketone + amine + [H] (NaBH₃CN or H₂/Pd).
Product: New C-N bond (amine).
e. Reduction of Nitrobenzene
Reagents: H₂/Pd or Sn/HCl.
Product: Aniline (Ar-NO₂ → Ar-NH₂).