cancer genetics since midterm
1/148
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
149 Terms
L#13: Studying cancer-causing mutations: model organism
Outline uses of cell culture models in cancer research
cells in culture
RNAi/CRISPR screens
library of gRNAs
lentivirus → deliver DNA to human cells
each cell deletes a dif gene in genome
seq to find which gRNA represented (↑,↓, =)
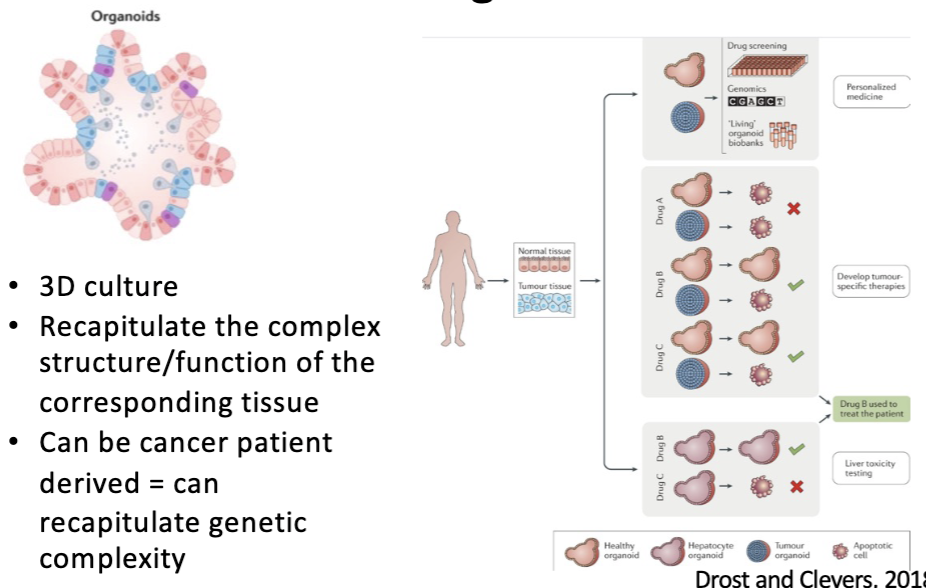
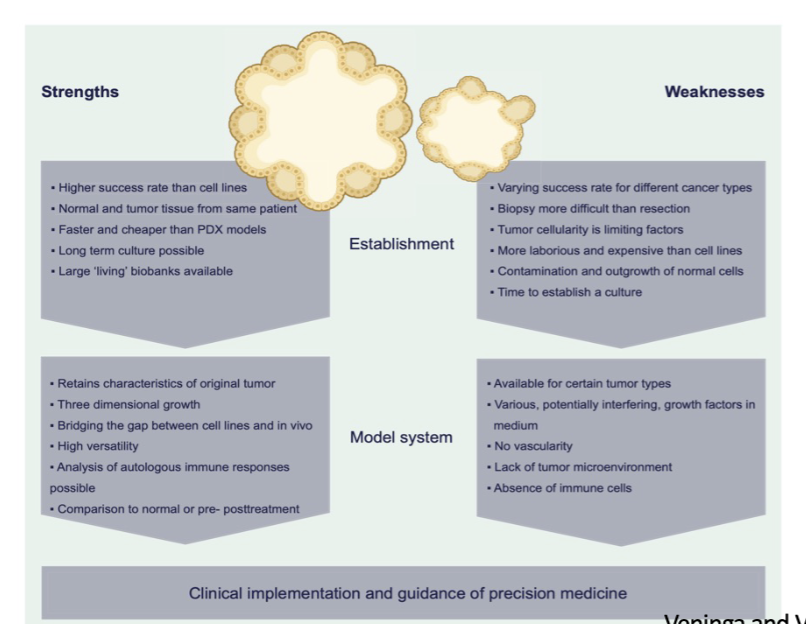
tumor organoids
3D culture
Recapitulate the complex structure/function of the corresponding tissue
Can be cancer patient derived = can recapitulate genetic complexity
↑ success rate, better than mice
no immune cells
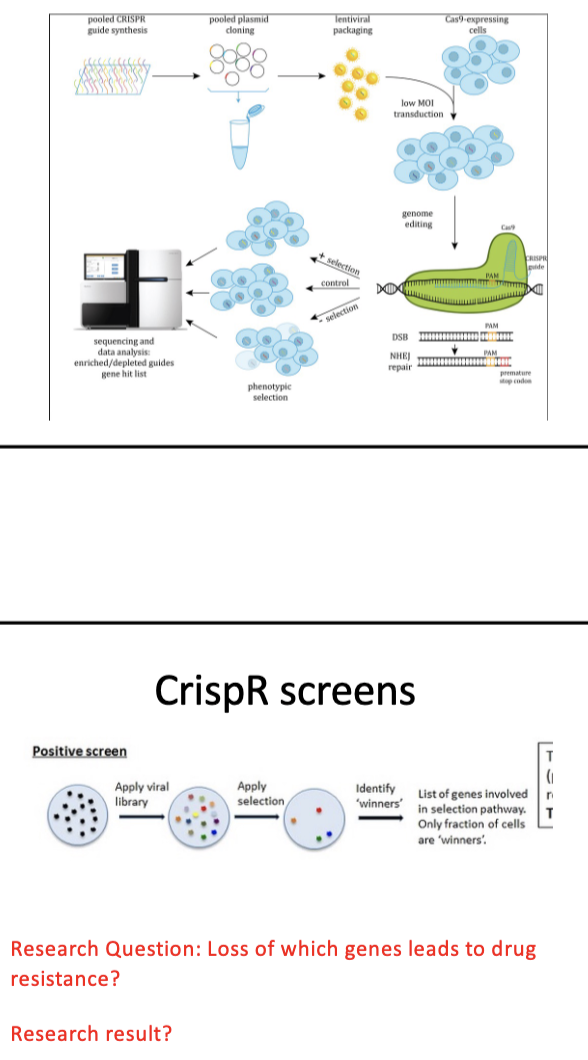
Outline uses of cell culture models in cancer research:
RNAi/CRISPR screens
RNAi
siRNA complementary to downregulanting the seq you want
leads to gene silencing (not completely eliminating gene function)
knock DOWN
CRISPR
library of gRNAs
lentivirus → deliver DNA to human cells
each cell deletes a dif gene in genome
seq to find which gRNA represented (↑,↓, =)
positive screen
essential for growth → KO → cell wont grow
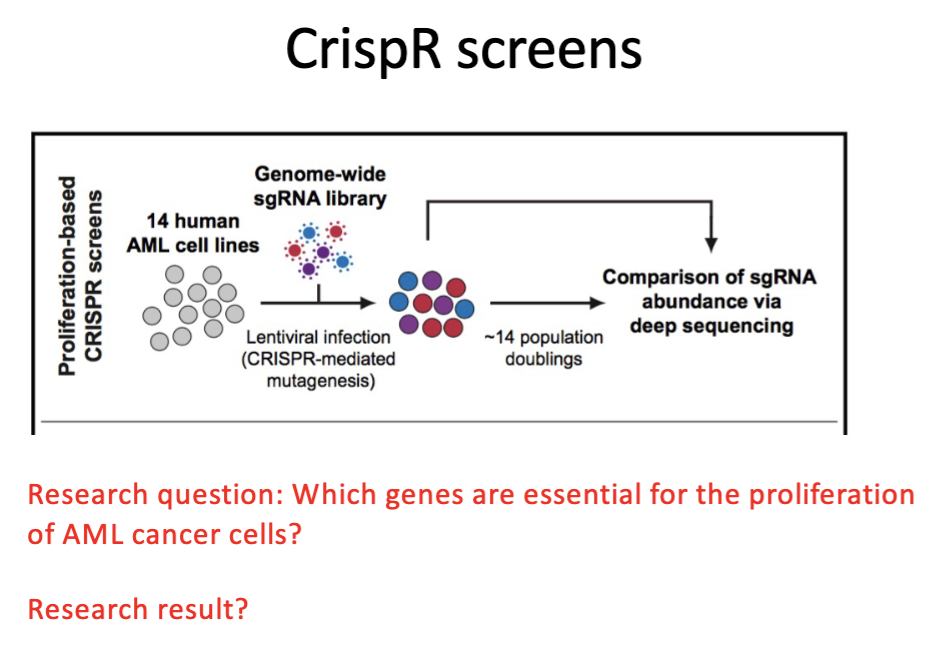
Outline uses of cell culture models in cancer research:
human organoids
3D culture
Recapitulate the complex structure/function of the corresponding tissue
Can be cancer patient derived = can recapitulate genetic complexity
↑ success rate, better than mice
no immune cells
L#13: Studying cancer-causing mutations: model organism
Discuss the use of model genetic organisms in cancer research.
Which model organisms have contributed?
mouse, rat, guinea pig, rabbit
worm, fly, yeast (genes)
zebrafish (immune)
Homolog
ortholog - human + c. elegan a-tubulin
paralog - within species (human has 10 copies, elephant has many copies of p53)
Forward genetics vs reverse genetics
forward - create random mutations → look for phenotypes
reverse - want to know a gene, what gene does what, seq → why is this mut associated with cancer
Why use mice in cancer research?
99% of genes have human counterpart
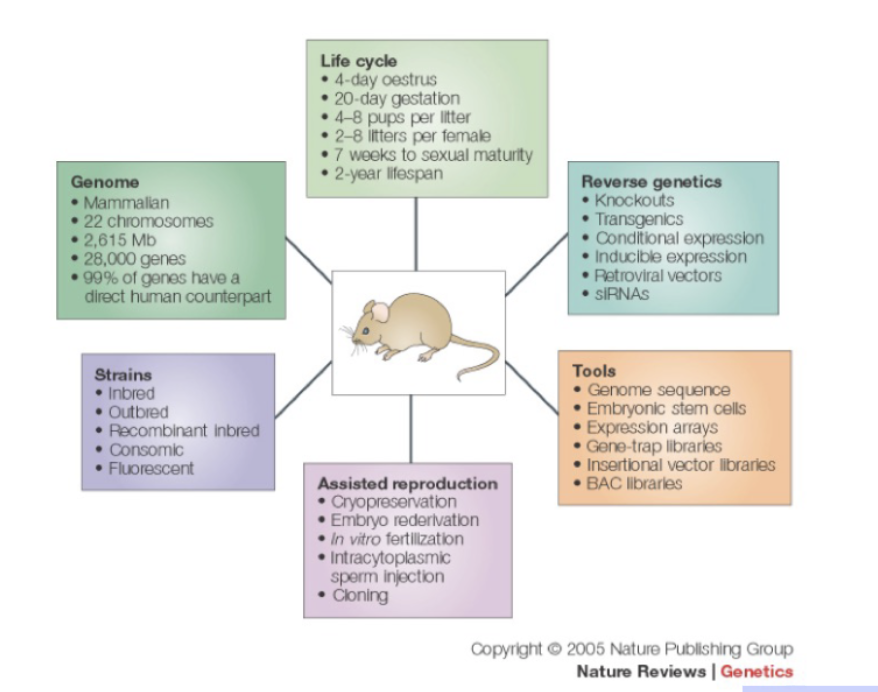
Studying gene function in mice
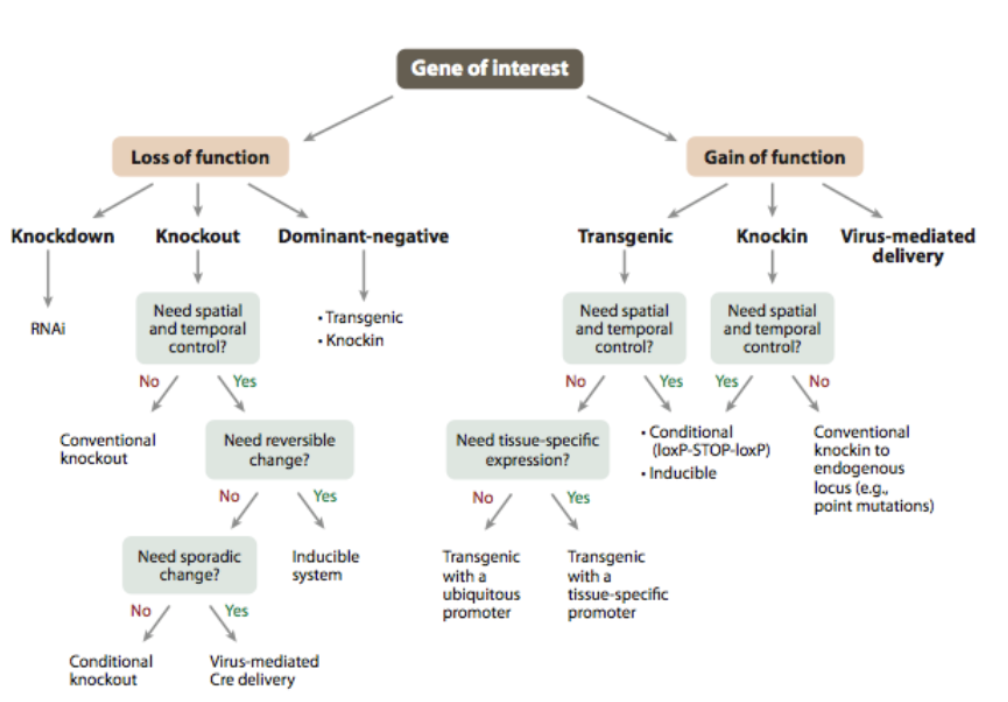
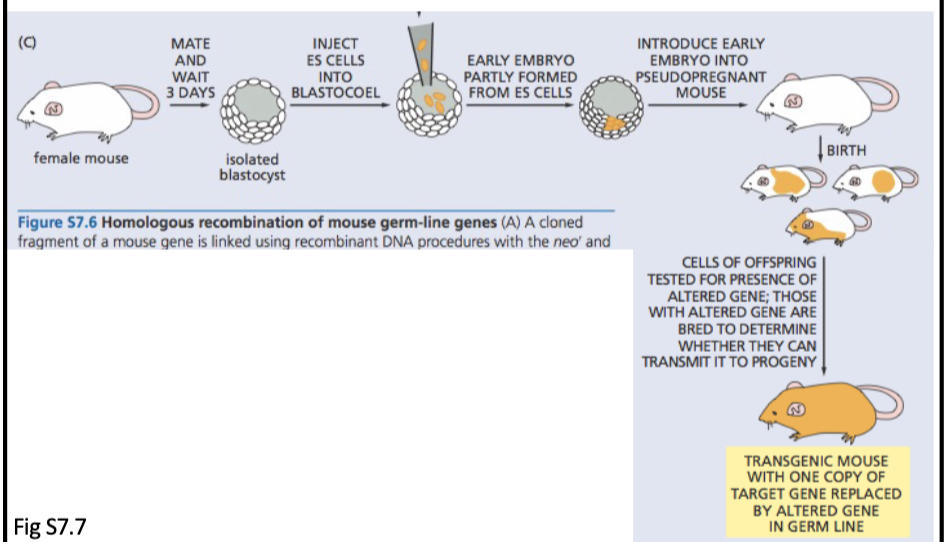
L#13: Studying cancer-causing mutations: model organism
Compare/contrast traditional approaches to making knockout/knock-in mice with use of crispR-cas9 system
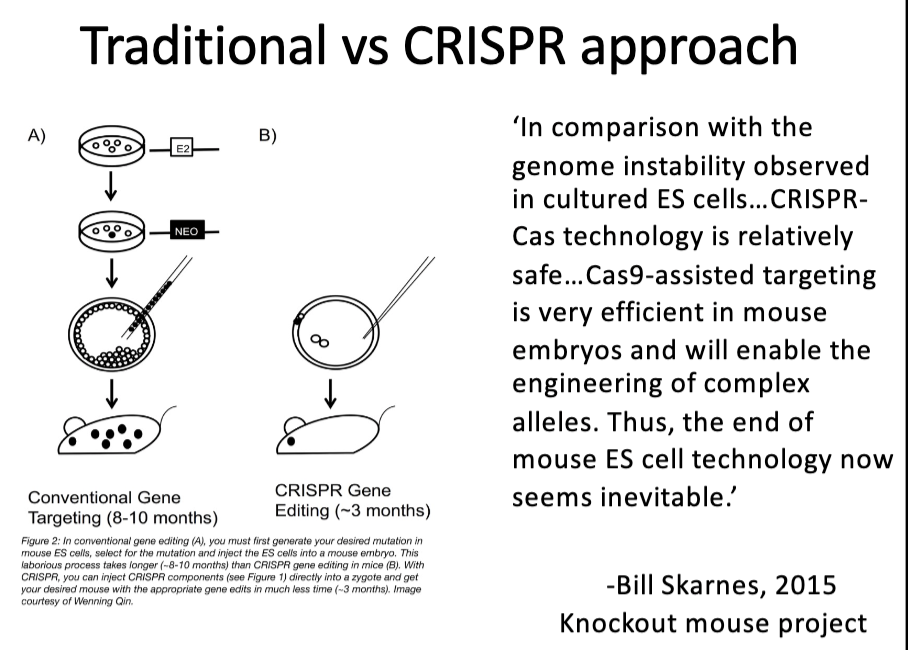
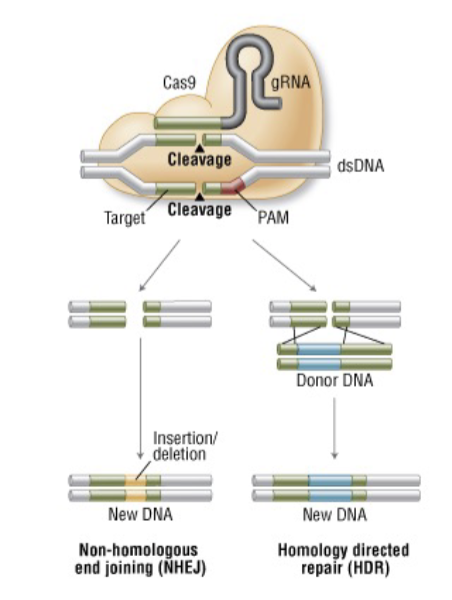
CRISPR/Cas9: Genome Editing
What types of changes can be made?
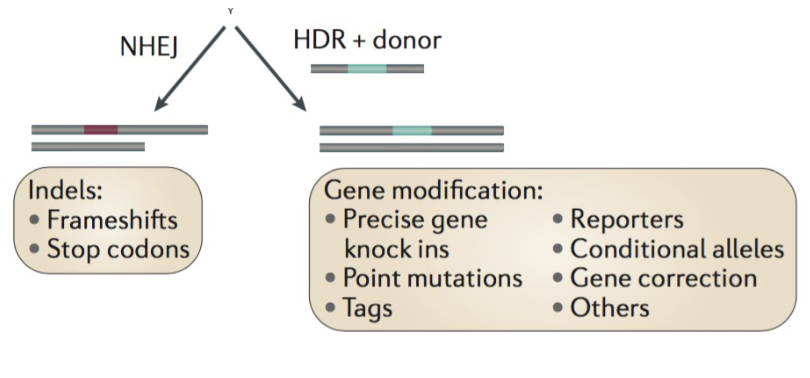
CrispR-cas-9 injection produces
______ results
various genotypic
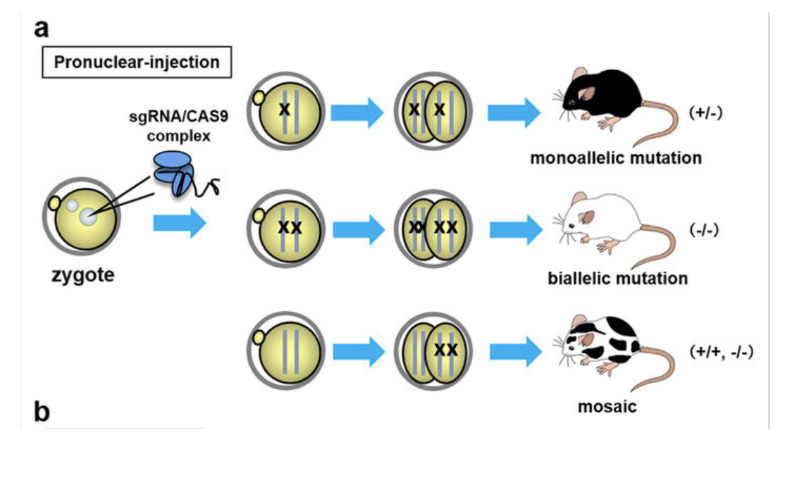
Controls, controls, controls...
Potential problems:
Mosaicism in founder
Off-target effects
Upregulation of homologous (unmodified) genes
Potential reactivation of gene product via use of alternative start, splicing out of premature stop etc.
Suppressor mutation in a second gene
L#13: Studying cancer-causing mutations: model organism
Explain why inducible systems may be used and how they work.
How can we study essential genes?
knockdown
tet off
tet on
Some knockouts aren’t viable

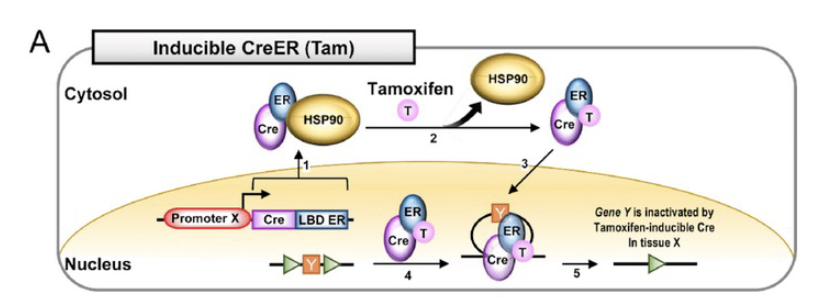
Cre-lox in practice
Diagram the 2 constructs required to make a mammary-specific deletion of BRCA-1 exon11.
conditional KO
promotor + CRE (control expression in certain tissues)
loxP sites + GOI
CRE induces recombination
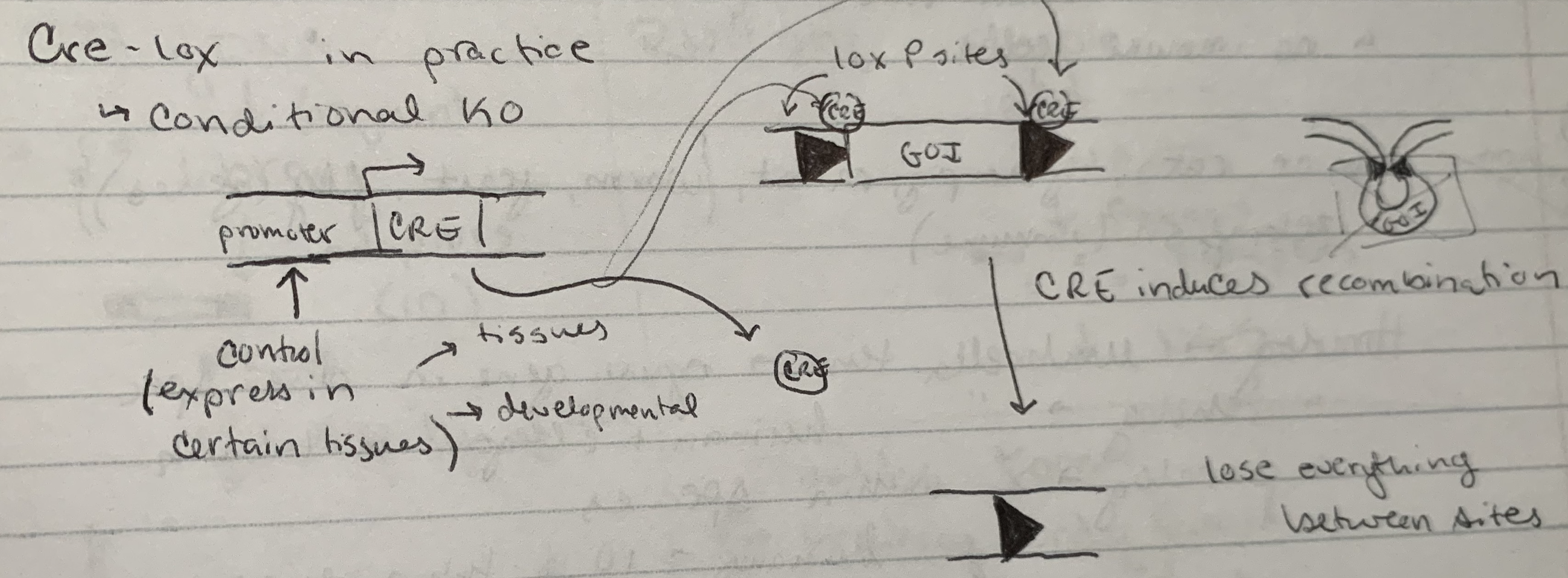
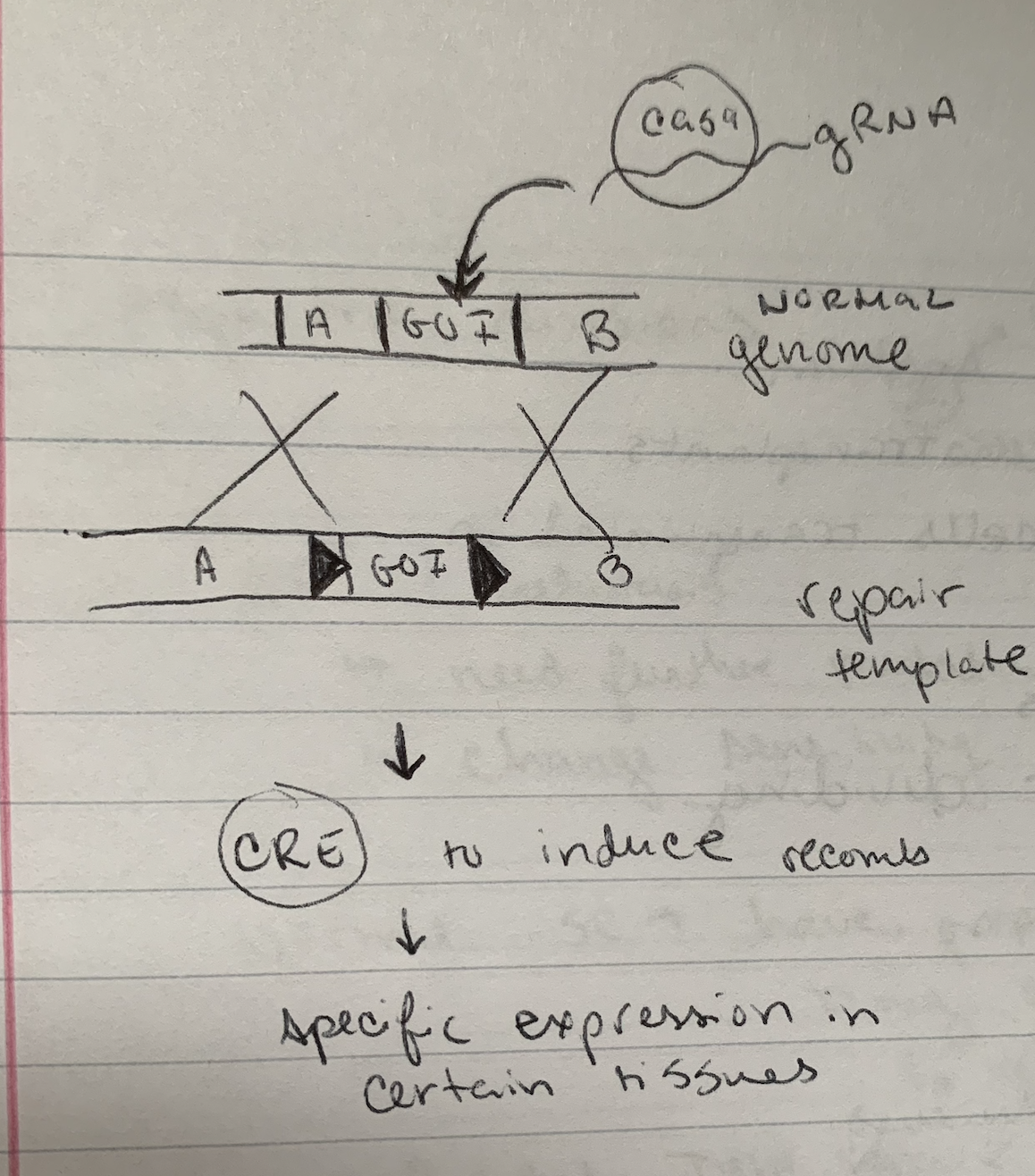
how to put in loxP sites
CRISPR + repair template
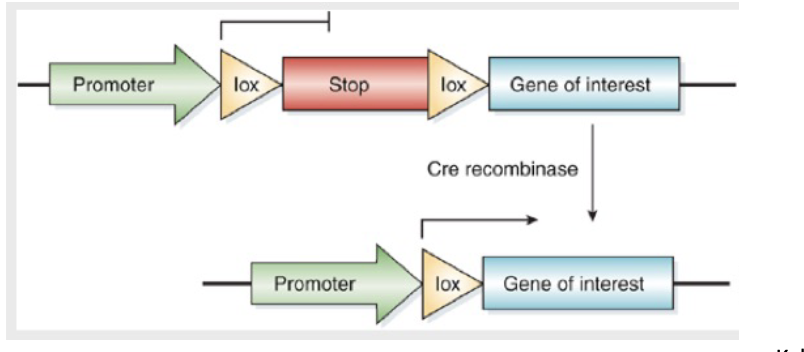
Variations on using the CRE-LOX system
If cre was expressed under control of a liver- specific promoter, what would this system allow?
not liver → no GOI protein
liver → protein of interest
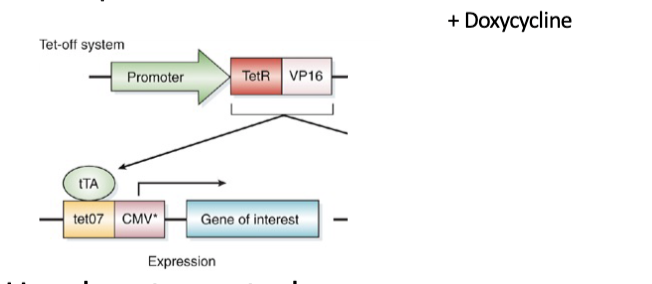
Tet off system
2 components
Use drug to control expression.
Dox present = off
Promoter gives spatial control
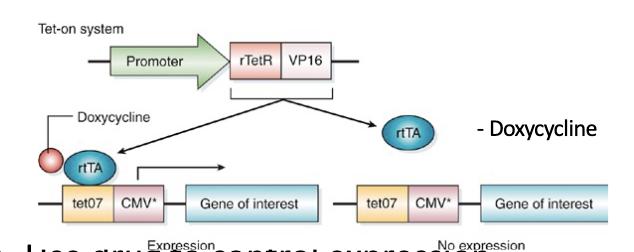
Tet on system
2 components
Use drug to control expression.
Dox present = on
Promoter gives spatial control
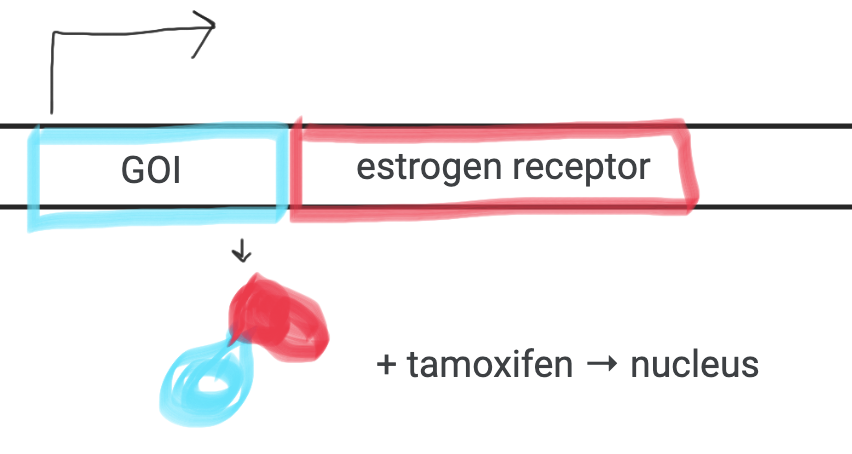
Tamoxifen-regulated
Use drug to control activity
add tamoxifen to ‘switch on’.
Promoter gives spatial control
Tamoxifen - used to control GOI getting into nucleus
(-) tam → outside nucleus
(+) tam → inside nucleus
L#13: Studying cancer-causing mutations: model organism
Design and interpret experiments that use model genetic organisms, including knockouts, knock-ins and conditionals.
L#14: Cancer stem cells
Explain self-renewal of normal stem cells, the role of transit amplifying cells and relate both to tissue homeostasis.
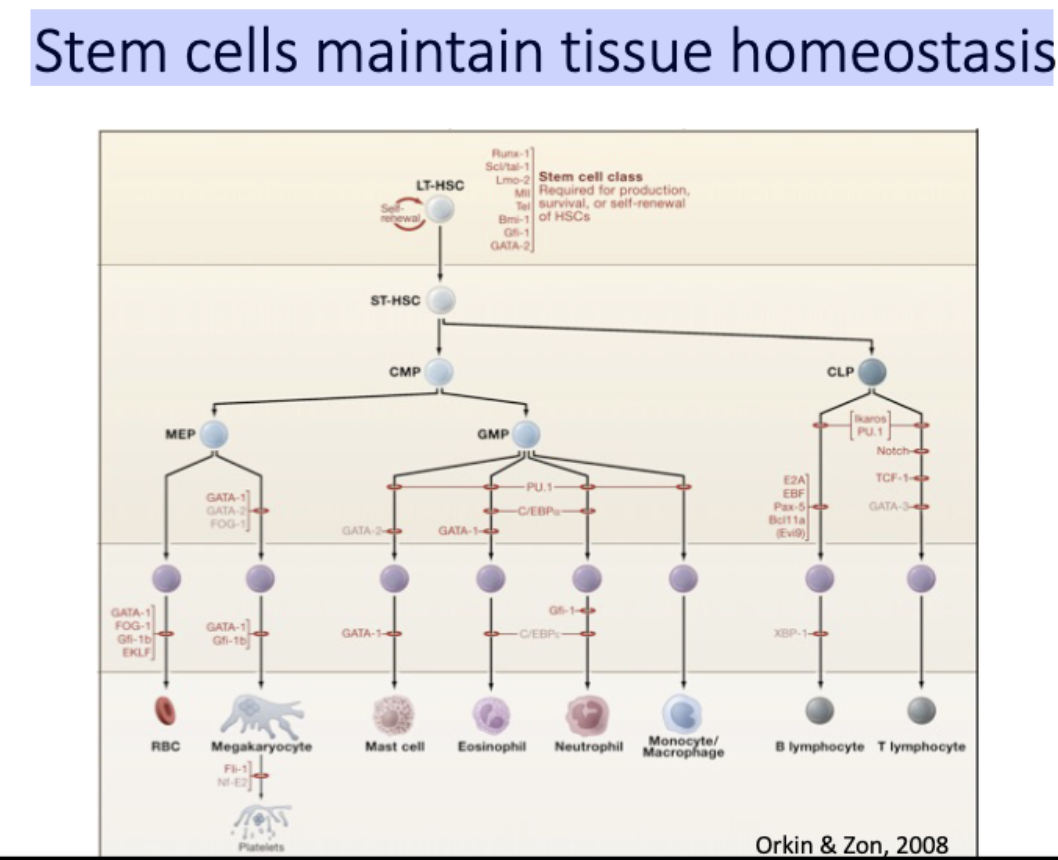
What is a stem cell?
An undifferentiated cell which has the ability to self-renew and to give rise to differentiated cells.
asymmetric division
A Stem cell maintains the ability to self-
renew over many divisions
daughter cells have different fates
Stem cells maintain tissue homeostasis
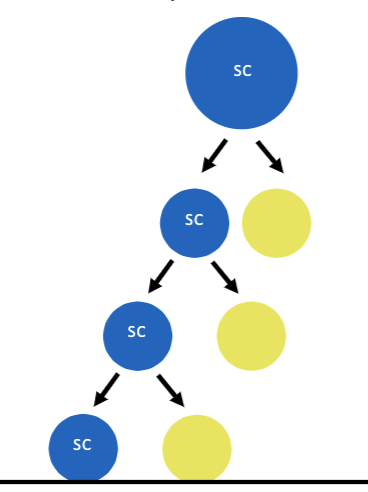
Transit amplifying cells
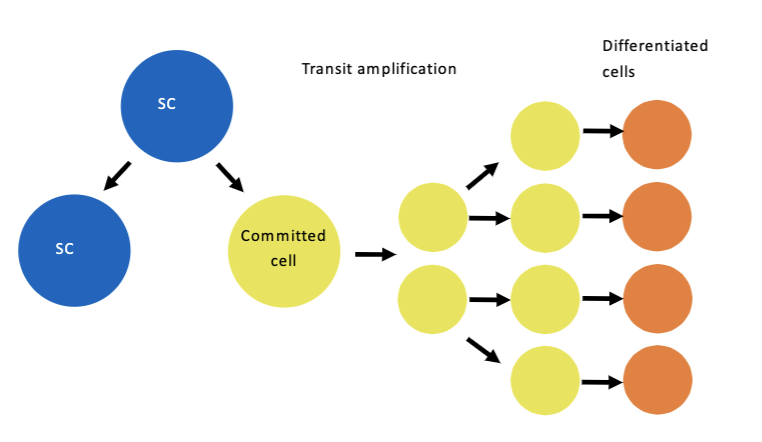
L#14: Cancer stem cells
Describe methods used to find CSCs and analyze data related to their use.
Are all tumor cells created equal?
Cells within a tumor show functional differences.
Only a subpopulation are capable of initiating tumors after transplant
= tumor propagating cells
= only 1% of tumour
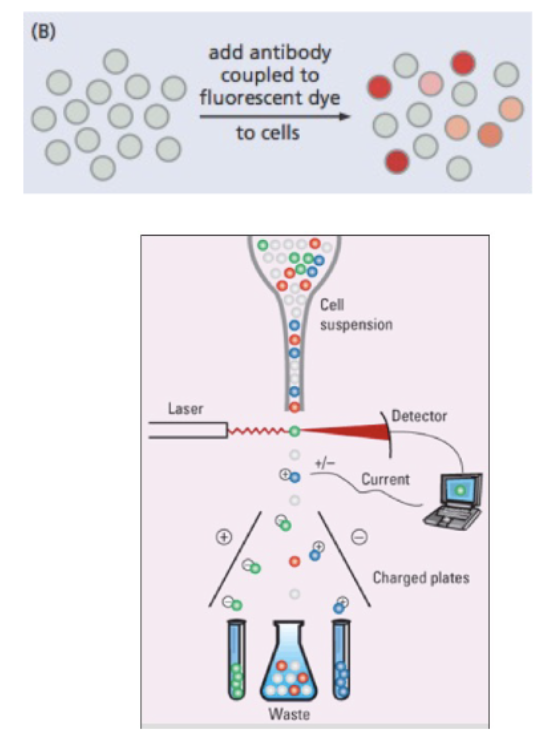
_____ can be used to separate cells based on surface markers
fluorescent activated cell signaling (FACS)
flow cytometry
Cell fractionation by FACS separates tumor propagating cells from bulk tumor cells
stain → antibody sticks to certain cells, color relate to proteins expressed on surface
laser → makes glow
detector
current → sorts
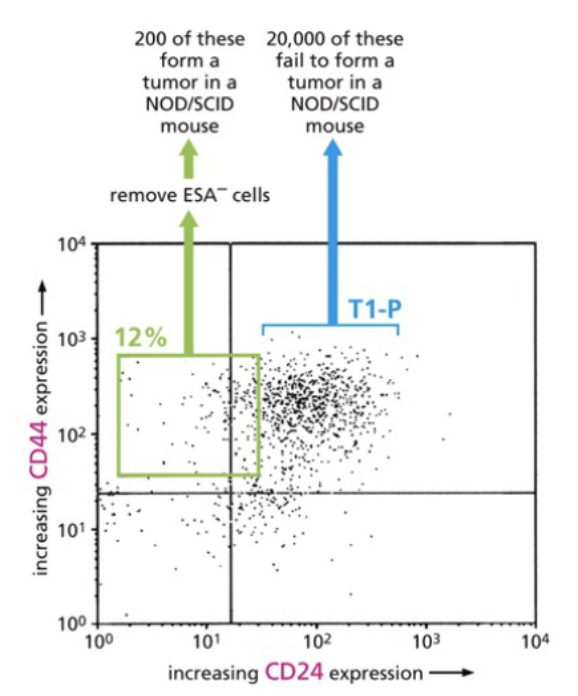
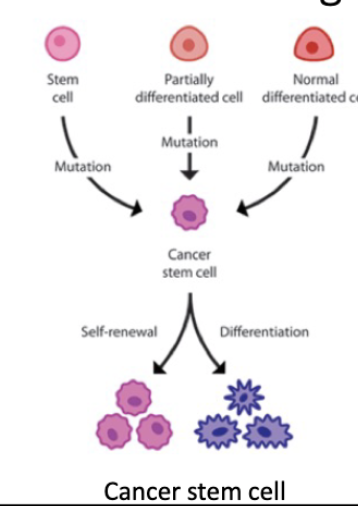
Cancer stem cell hypothesis
• A subpopulation of cells within the tumour has preferential capacity for driving tumor growth
• These CSCs promote and regrowth at a new site
• The CSCs can recreate tumor heterogeneity.
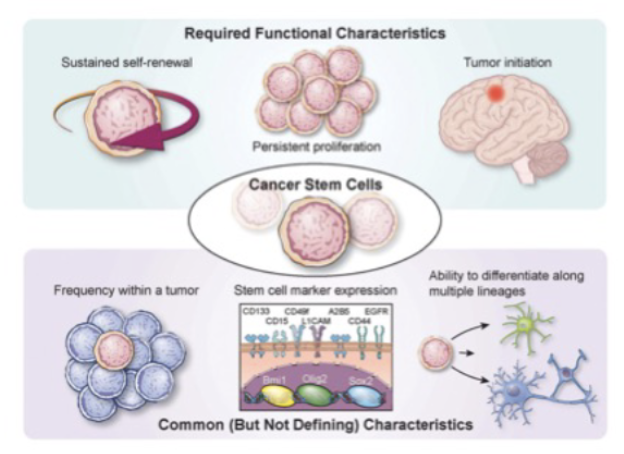
Cell surface markers ____ for CSC (but don’t purify them)
enrich
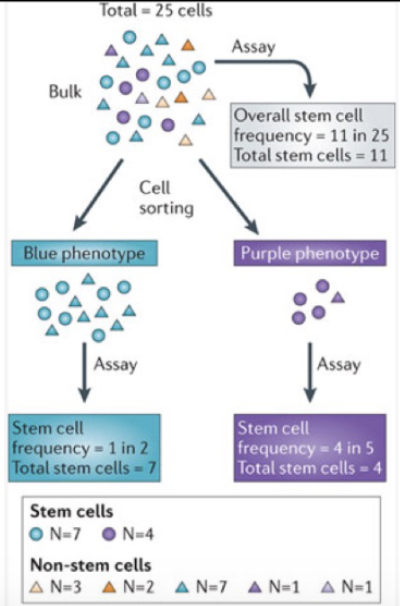
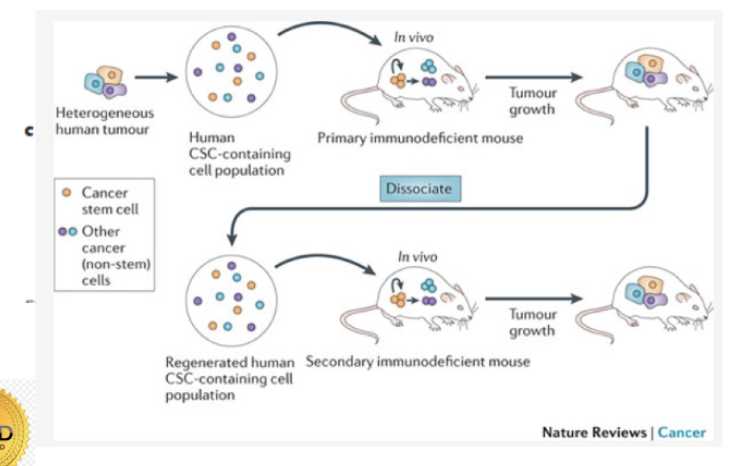
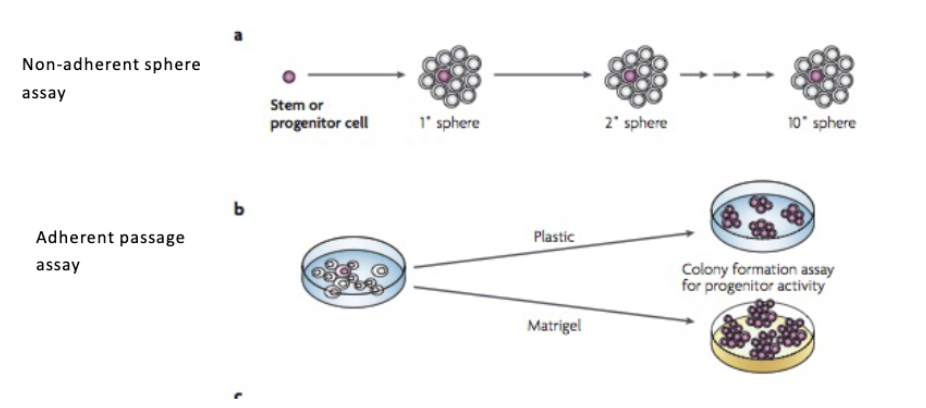
Identifying CSCs
colony forming assays
L#14: Cancer stem cells
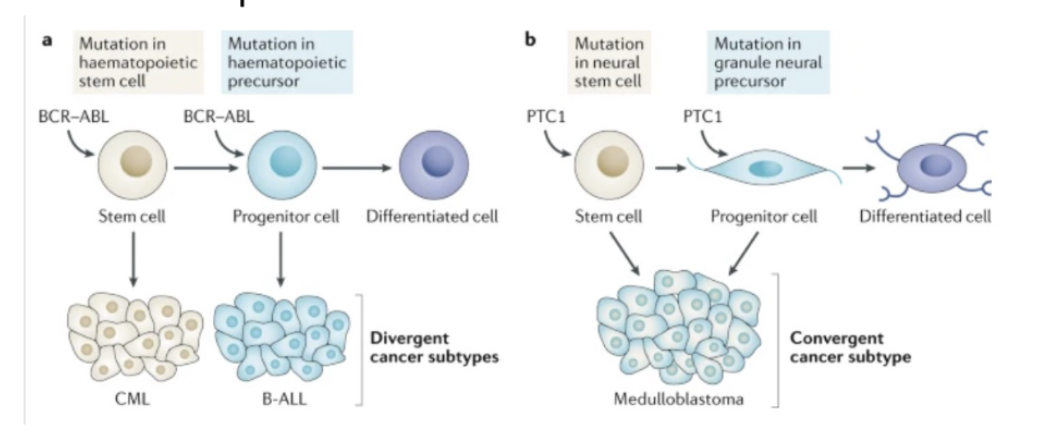
Discuss CSCs in the context of cell-of-origin.
What is the cancer cell of origin?
Impact of cell-of-origin on cancer can be context-specific
L#14: Cancer stem cells
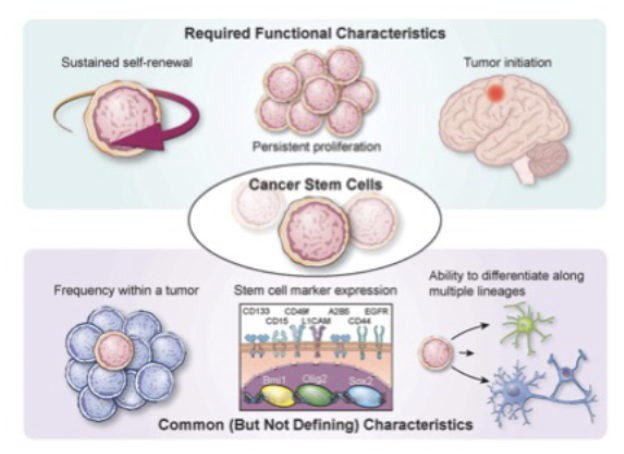
Describe the features of cancer stem cells. Compare and contrast normal stem cells and cancer stem cells.
normal vs csc
CSC dont reinvent the wheel
Cell surface markers
reactivate Signaling pathways
Asymmetric divisions
EMT
Normal stem cell signals activated in CSCs
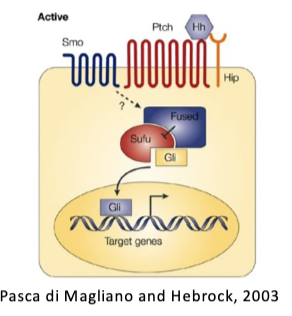
WNT, NOTCH, HHG signaling pathways active in stem cells AND often mutated (to activate) in cancer cells
Colon cancer: APC mutation ->activated WNT
Medulloblastoma: SMO, PTCH1, or Gli1 mutation -> Hhg signalling
T-ALL: NOTCH mutations
Signals serve as drivers. i.e. stem cell signals are oncogenic
Dysregulated asymmetric cell divisions
Cell fate determinants are asymmetrically inherited in normal stem cell divisions.
Dysregulation of asymmetric cell divisions can lead to increased self-renewal.
asymmetric localization of proteins can inc SC in tumor
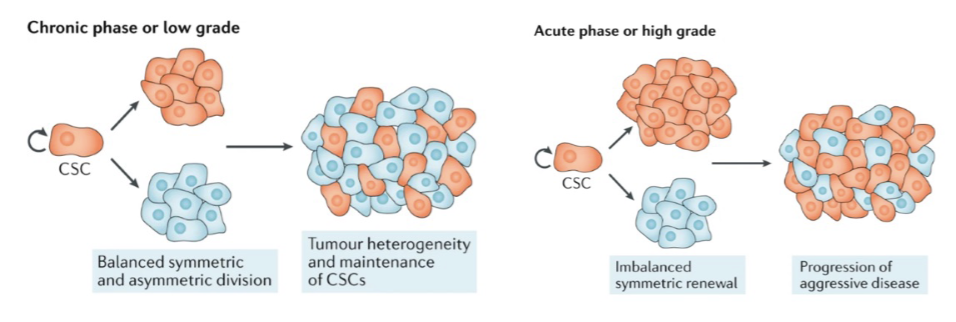
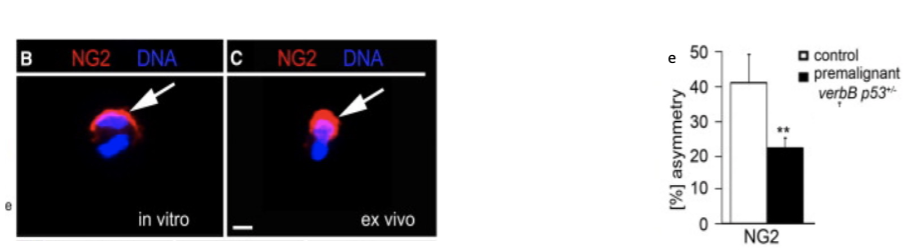
Asymmetric Cell division in oligodendrocytes
Studies in mouse models have shown that oligodendrocyte progenitor cells (OPC) carrying tumor-initiating
mutations are a cell of origin of oligodendroglioma, a progressive primary malignancy of the adult central
nervous system and major glioma class.
Fig. B&C dividing non-cancer cells labelled for the proteoglycan, NG2. E) Asymmetric divisions in WT and a
mouse model of glioma were compared. Modified from Sugiarto et al, 2011.
L#14: Cancer stem cells
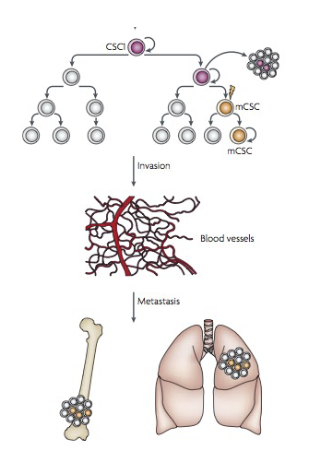
Discuss the implications of the existence of cancer stem cells, for tumor initiation, metastasis and therapy resistance.
• Cells that seed metastases are CSCs
• Are all CSCs capable of metastases?
OR
• do they need to evolve new abilities to undergo metastases?
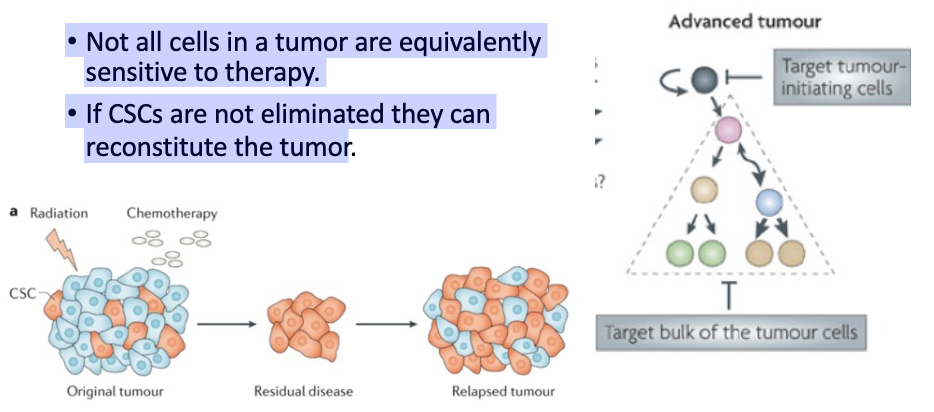
CSCs and treatment.
• Not all cells in a tumor are equivalently sensitive to therapy.
• If CSCs are not eliminated they can reconstitute the tumor
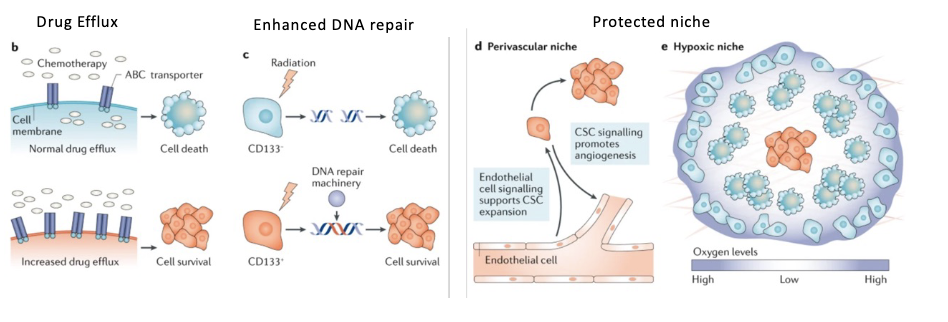
(normal) SC properties acquired by CSCs provide an advantage in surviving chemotherapy
normal SC have properties that make long lived
drug efflux (dont want to be killed by random toxins)
good for cancer
efficient dna repair
protected niche → buried
Exploiting CSC properties in treatment
• ‘Differentiation therapies’ in development.
push toward dif = no longer sustain tumor
• Eg SMO antagonists to block Hhg pathway approved for advanced basal cell carcinoma
• Other pathway antagonists in development.
L#14: Cancer stem cells
Describe factors that aid CSCs therapy resistance.
L#15: Insensitivity to anti-growth signals
Discuss cancer-inducing mutations with reference to inheritance (dominant/recessive). How can this be determined experimentally?
insensitivity to antigrowth signals
Two major routes to insensitivity to antigrowth signals
• Pro-growth signals override normal controls
• Loss of antigrowth signaling components (TS)
Tumor suppressor - definition
A gene whose product functions in controlling cell growth
AND
Whose LOF can drive tumorigenesis
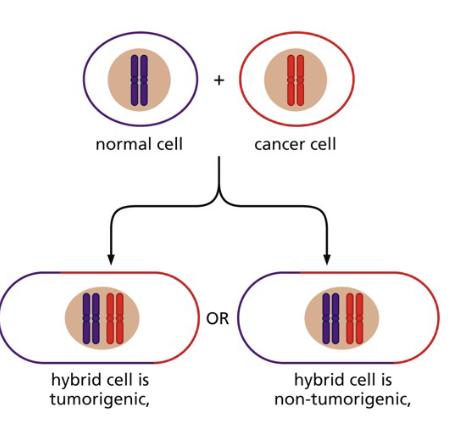
Are cancer-inducing mutations dominant or recessive?
normal + cancer → hybrid
~ phenotypes
remains tumoriogenic = dominant
not tumoriogenic = recessive
If a cancer-inducing mutation is recessive what does it mean for a cancer cell genotype?
homozygous (cancer cell genotype of recessive mutation)
2 identical (mut) alleles
2 different (mut) alleles
hemizygous
1 mut allele
1 deletion
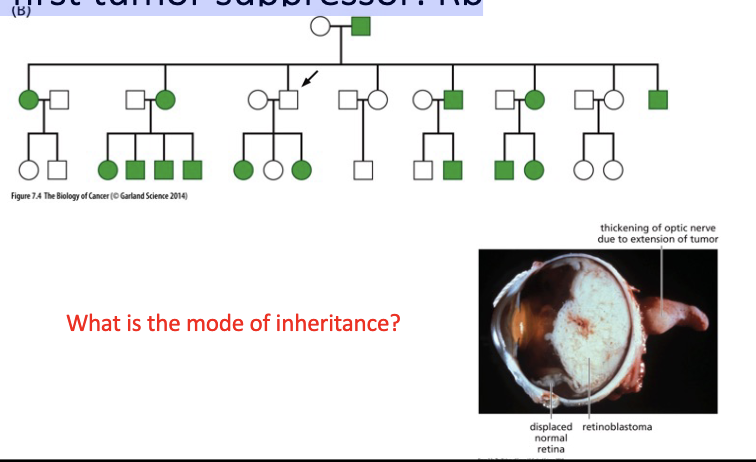
The first tumor suppressor:
Rb
L#15: Insensitivity to anti-growth signals
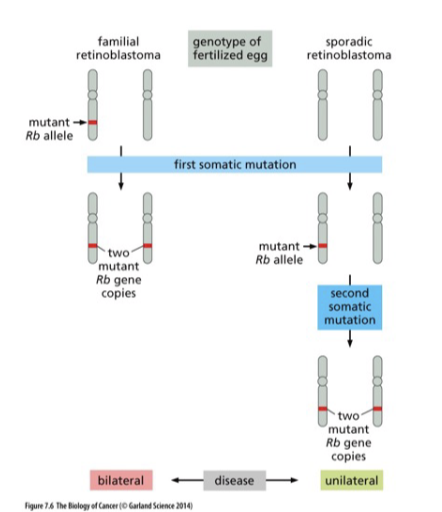
Describe knudson’s 2 hit hypothesis.
L#15: Insensitivity to anti-growth signals
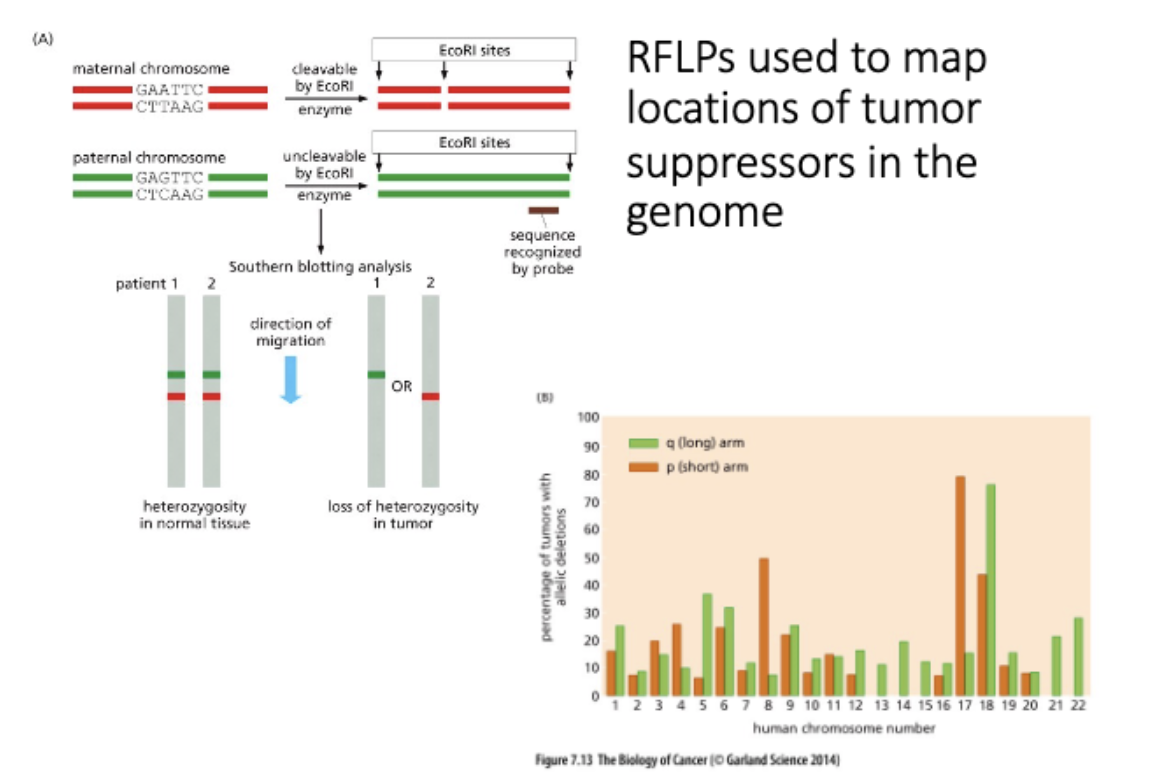
Explain what LOH is and interpret data related to detection of LOH.
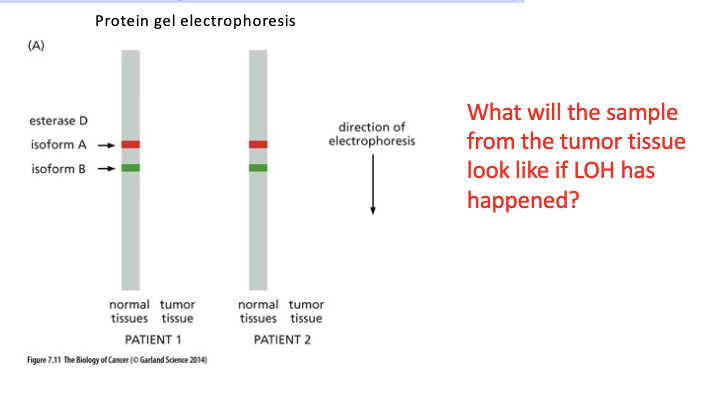
Direct testing of the LOH theory
L#15: Insensitivity to anti-growth signals
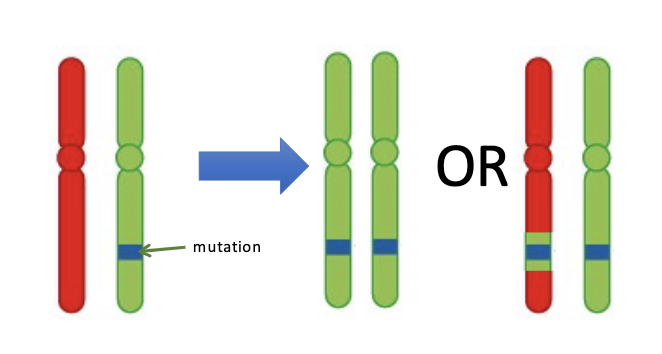
Diagram models of how LOH arises.
1. Mitotic recombination
2. Gene conversion (strand switching during replication)
3. Chromosomal structural changes – hemizygosity
4. Non-disjunction and subsequent chr loss
LOH arises
mitotic recombinaton
dont expect recomb in mitosis
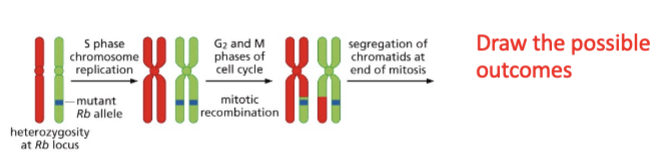
LOH arises
gene conversion
template switching by DNA pol
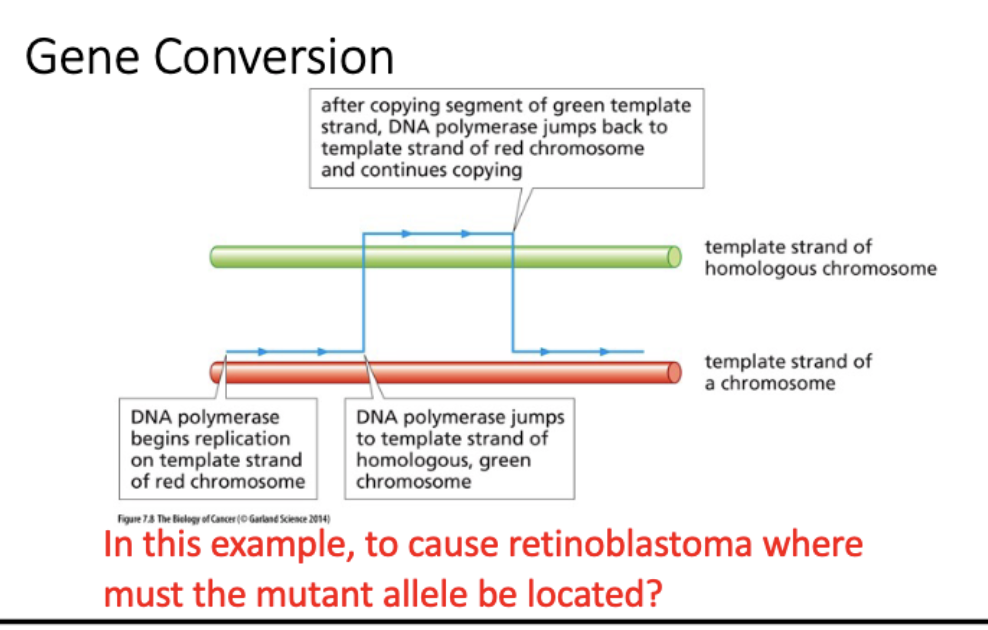
LOH arises
Non-disjunction followed by chromosome loss
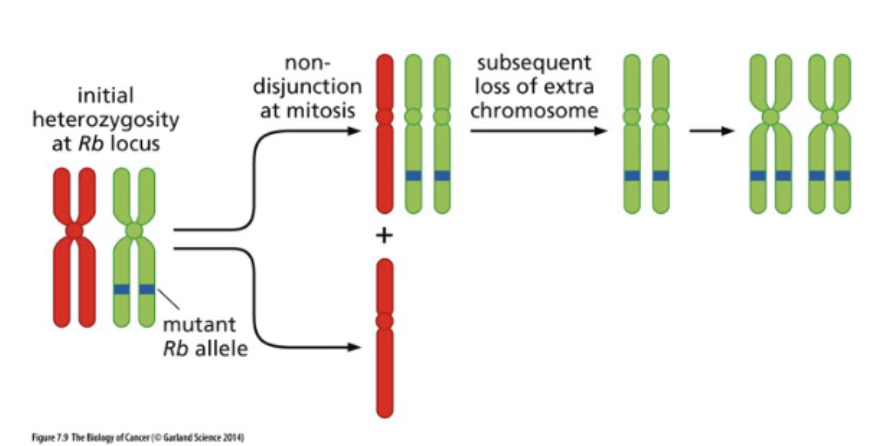
LOH arises
Chromosome Structural Changes
= hemizygosity
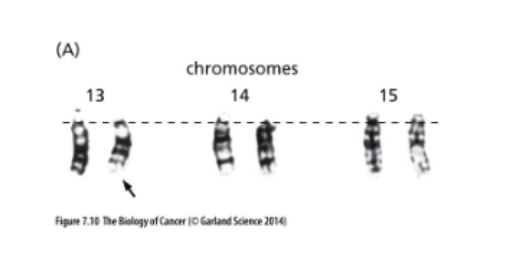
L#15: Insensitivity to anti-growth signals
Describe non-mutagenic pathways that can lead to gene inactivation and how they can be monitored experimentally
Non-mutagenic processes -RNA
Premature polyadenylation.
Promoter methylation can inactivate ts genes – crosstalk with chromatin modifications.
EVIDENCE:
CpG islands in promoters – increased me in cancer
CIMP – CpG island methylator phenotype can be associated with increased DNMT3B expression.
Me promoters show LOH
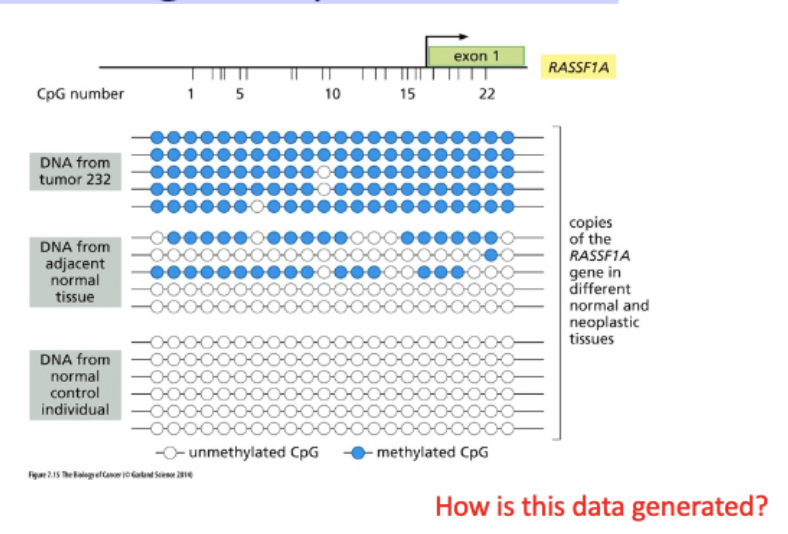
Monitoring methylations status
BS seq
nanopore seq - direct detection
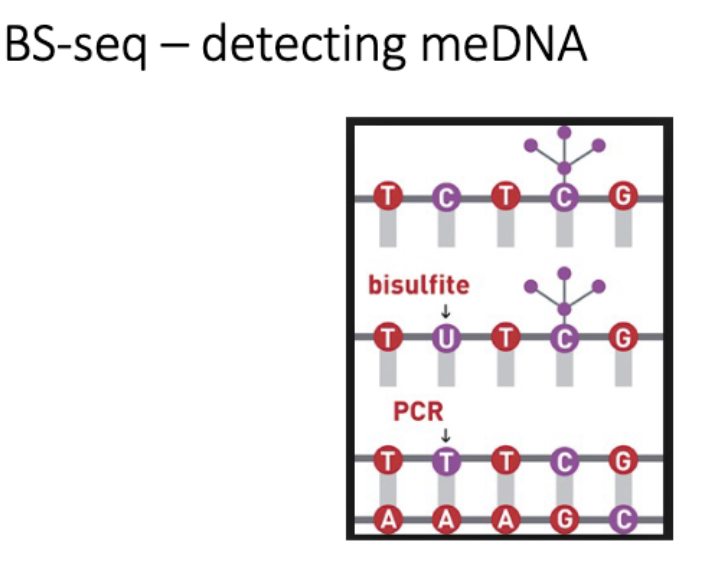
L#15: Insensitivity to anti-growth signals
Discuss the network of interactions that underlies cell cycle entry.
Functions of tumor suppressors
Unifying feature:
Operate to decrease likelihood of cancer development
Regulate circuits that govern cell proliferation and survival
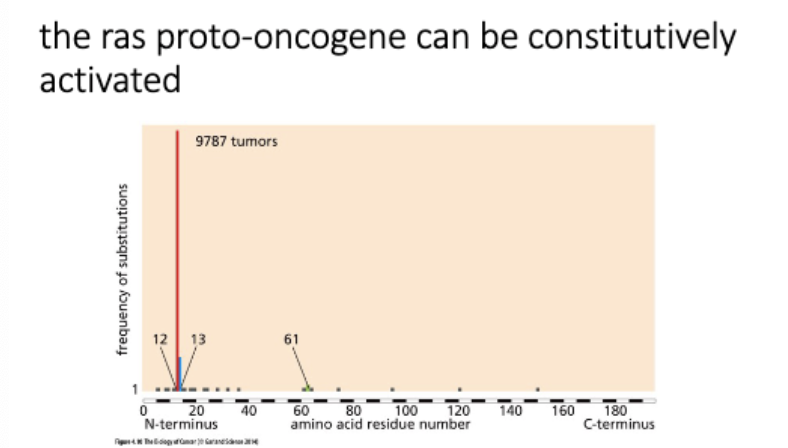
Tumor suppressors often involved in negative feedback:
NF1
switches off at end
Ras → small GTPase, downstream signaling
ACTIVE - Ras+GTP
signals grow + divide
Ras GAP - GTPase activating protein NF1 mut = Ras stuck in active form
INACTIVE - Ras+GDP
Ras GEF - GTP exchange factor
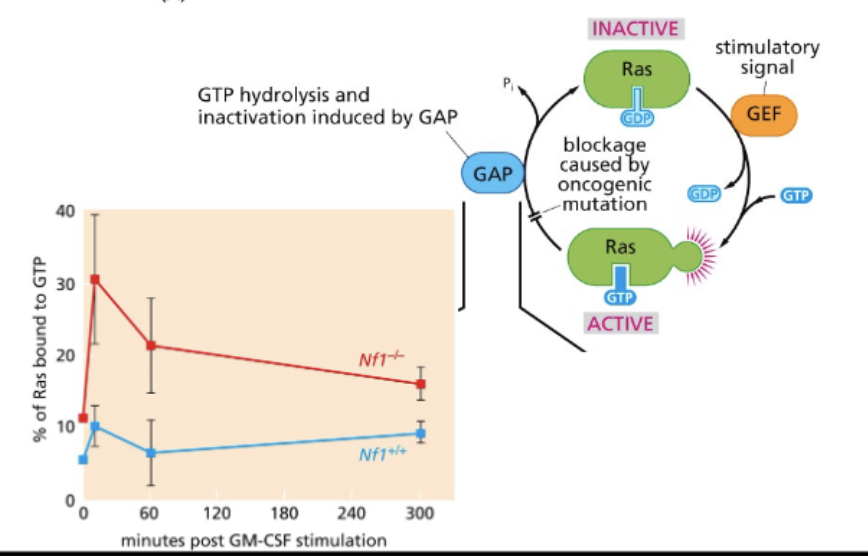
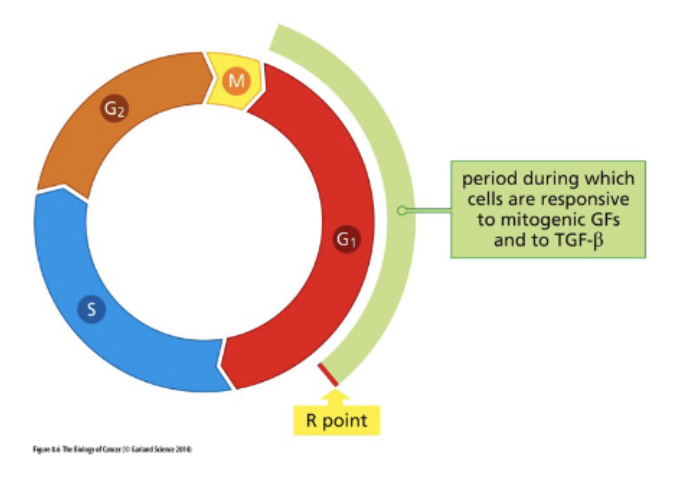
Focus on cell cycle entry
Rb regulates cell cycle entry
mitogenic - pro prolif signals
R point - point of no return
Mitogenic signal activates CDK
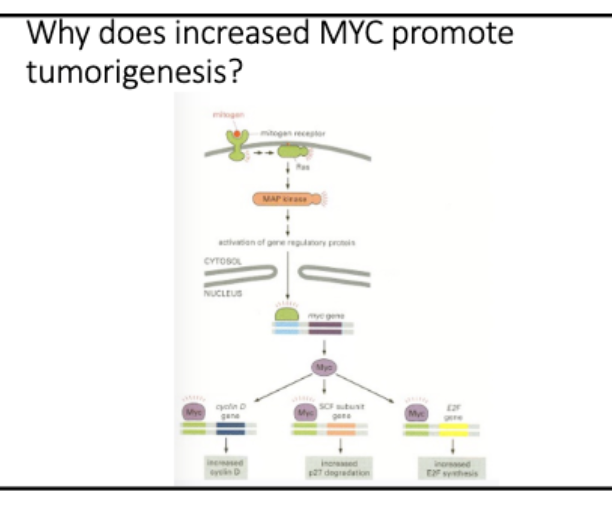
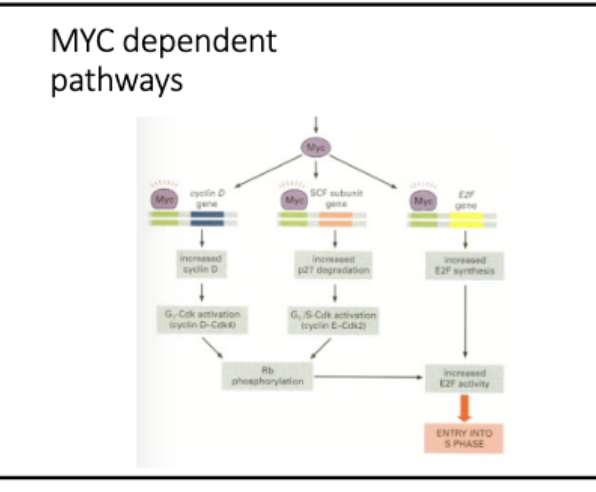
• Mitogen signaling activates Myc
• Myc is a TF
• Myc activates cyclinD-CK4/6
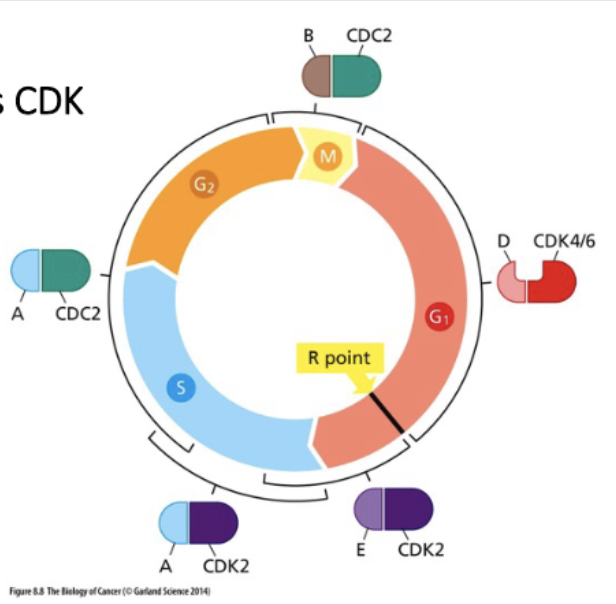
Cyclin D/CDK initiates Rb phosphorylation,
CyclinE/CDK continues
mittogenic signaling → myc (TF) → cyclinD+E → (P) Rb
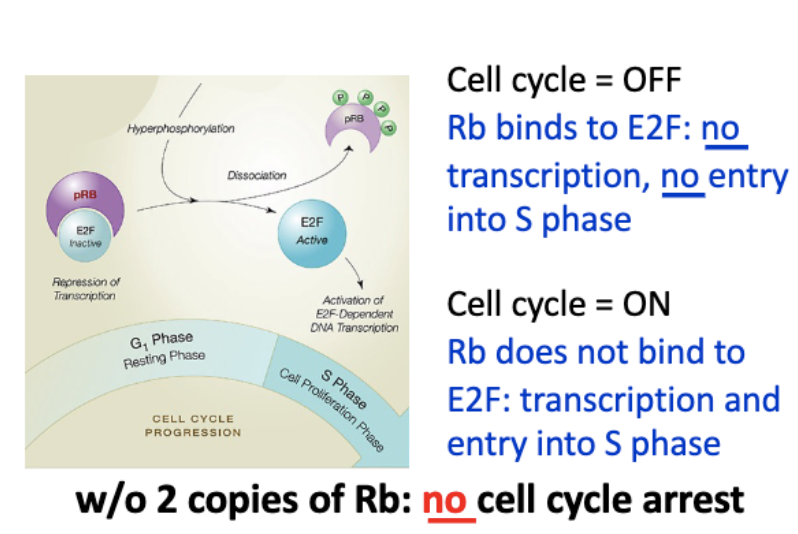
Rb - binds E2F → no transcription
↓ cyclin D+E
Rb (P) - does not bind E2F → active transc
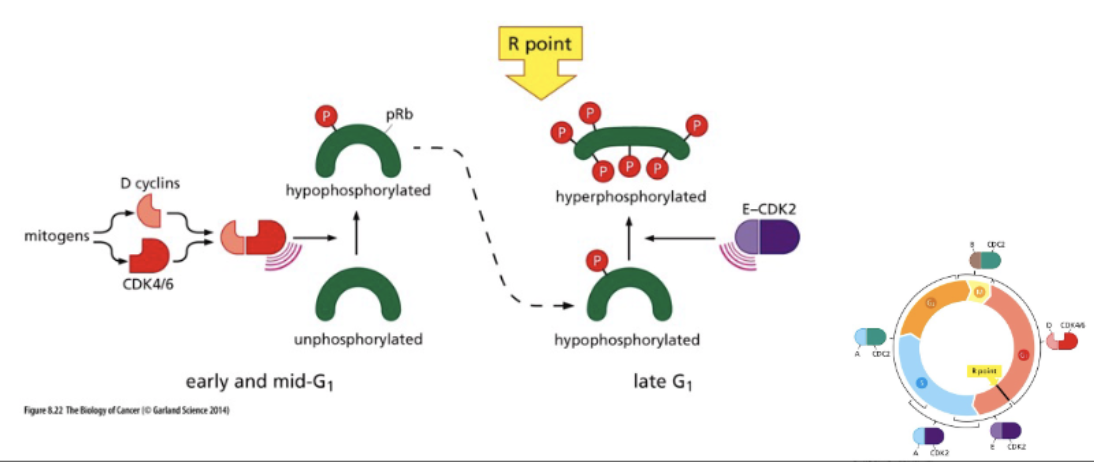
RB regulates progression through G1 phase
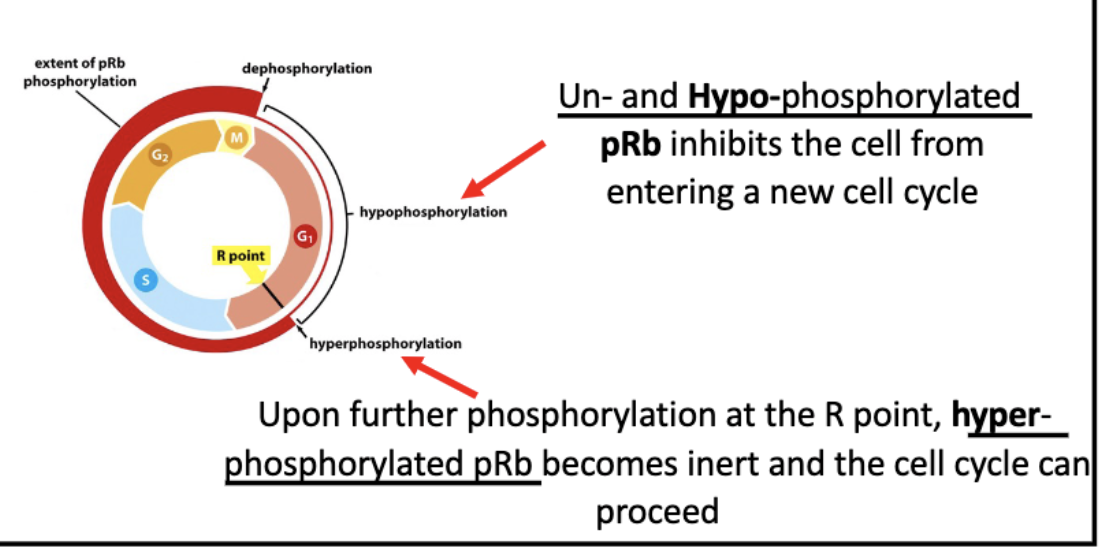
Rb regulates E2F activity
E2F = transcription factor of S phase proteins
bond to Rb = inactive
unbound = active transcription
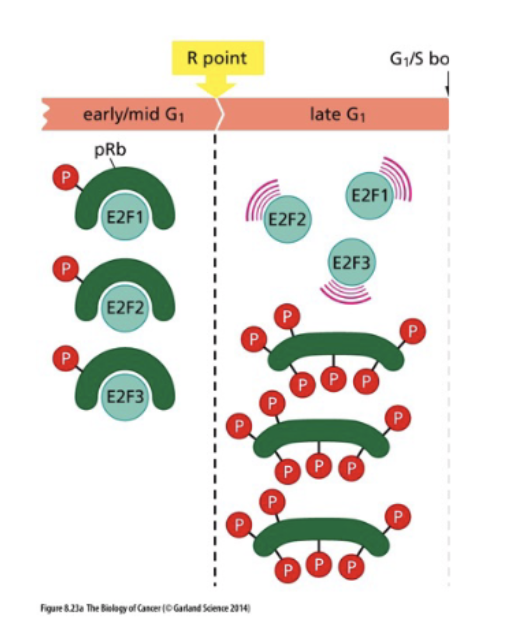
hyperphosphorylation of Rb = dissociation from E2F = transcription activation
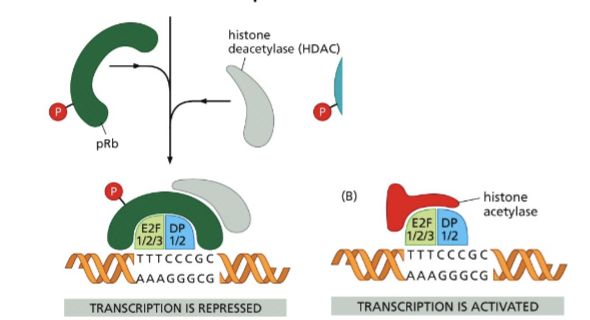
Rb, the retinoblastoma protein regulates the cell cycle
L#15: Insensitivity to anti-growth signals
Use information on gene function in the cell cycle to hypothesize the outcome of a given experiment.
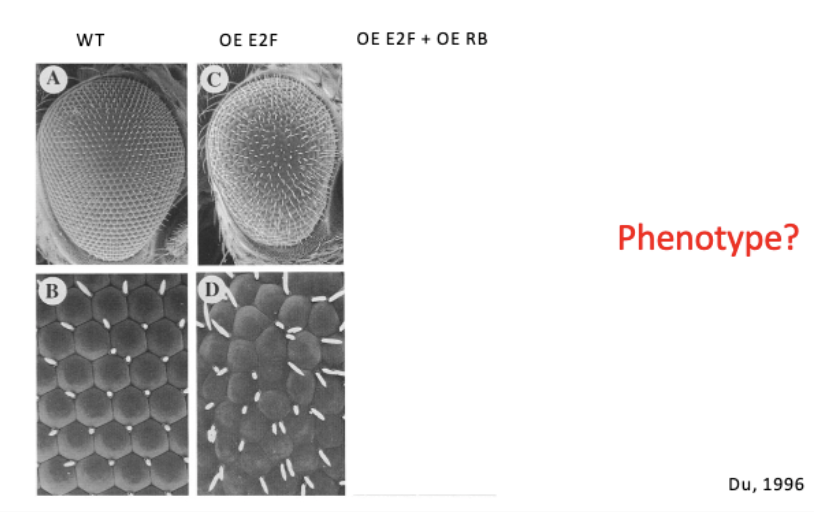
investigating a genetic interaction between Drosophila Rb and E2F
conditional KO - cre lox
overexpress E2F = ↑ cell cycle = rough eye
overexpress Rb = gets better

L#15: Insensitivity to anti-growth signals
Diagram methods used to study tumor suppressor function and interpret data stemming from their use.
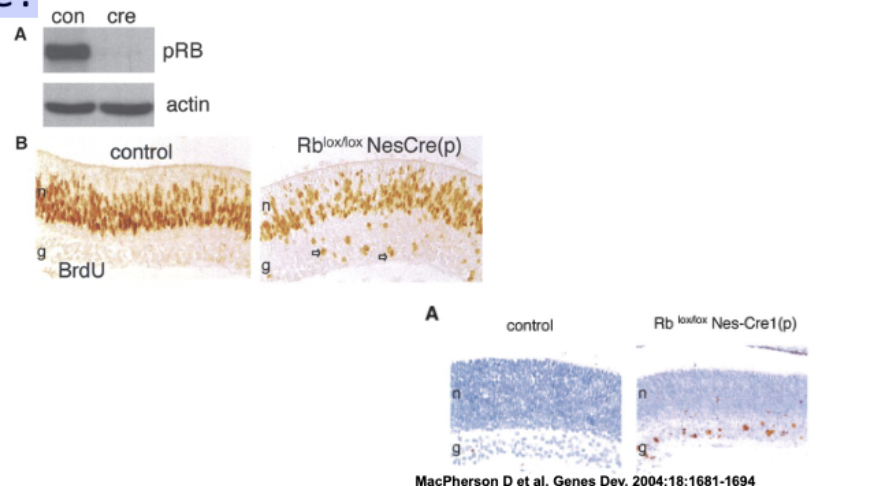
Does knockout of RB cause retinoblastoma in mice?
—nesprin eye prom—cre——————-lox-Rb-lox—
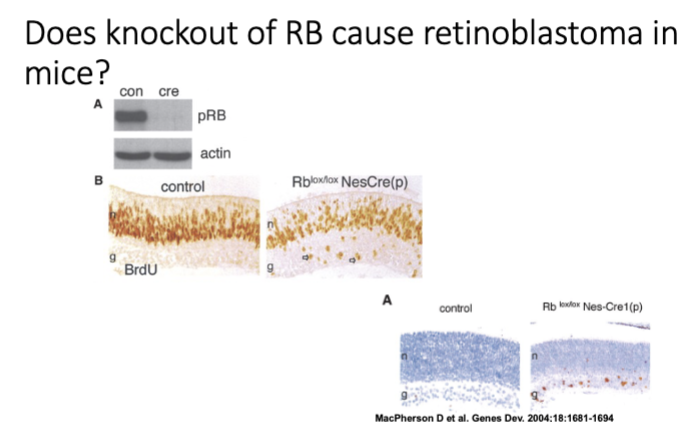
Does knockout of RB cause retinoblastoma in
mice?
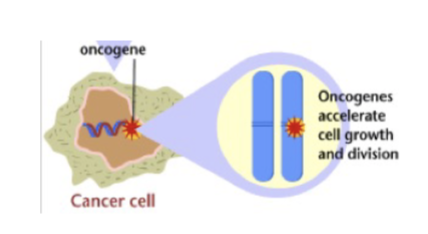
L#16: Increased growth signaling
Contrast proto-oncogenes and oncogenes.
Oncogene - definition
• A gene whose expression contributes to cancer. eg. pro-proliferation signal, Ras
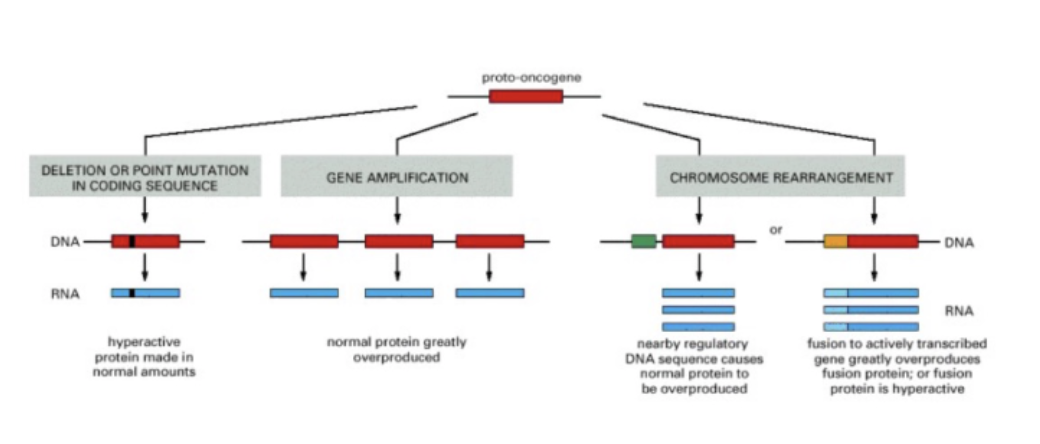
protooncogene → oncogene
how?
The path to oncogene
protonco → onco
change in coding seq - hyperactive protein (normal amounts, abnormal protein)
gene amplification - more copies of protein (normal protein, abnormal amount)
chr rearrangement - bring together seqs
Normal protein – more of it:
Gene amplification
Altered expression
Disruption of gene function
Gene fusion – novel hybrid protein
Deregulated activity – ligand independent signaling
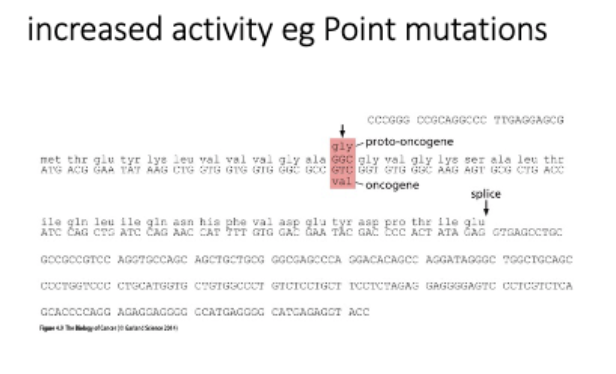
mutation = increased activity
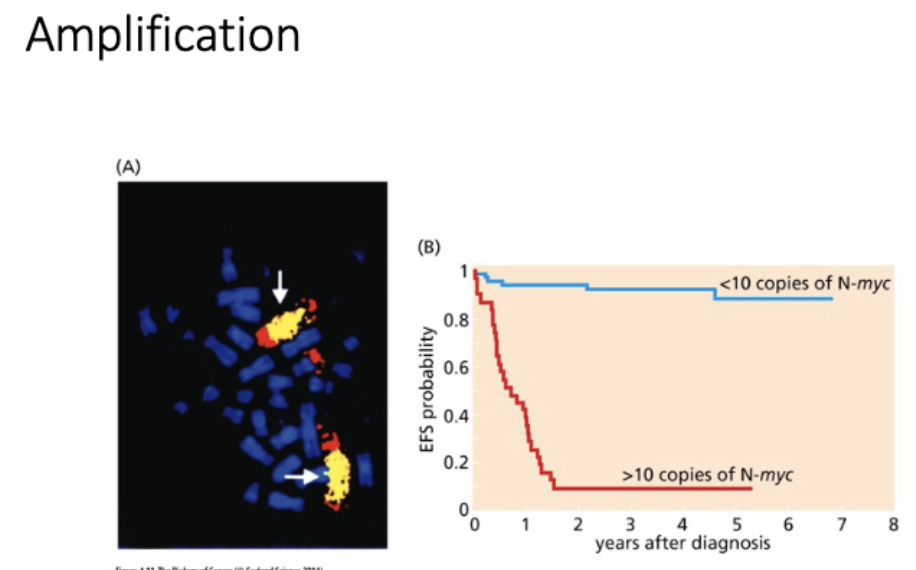
L#16: Increased growth signaling
Explain with examples how oncogene creation happens without changing protein function and diagram how it may come about.
gene amplification
homogeneously staining regions (tandom duplications in chr)
vs
double minutes
extra chr arrays - not under normal regulation
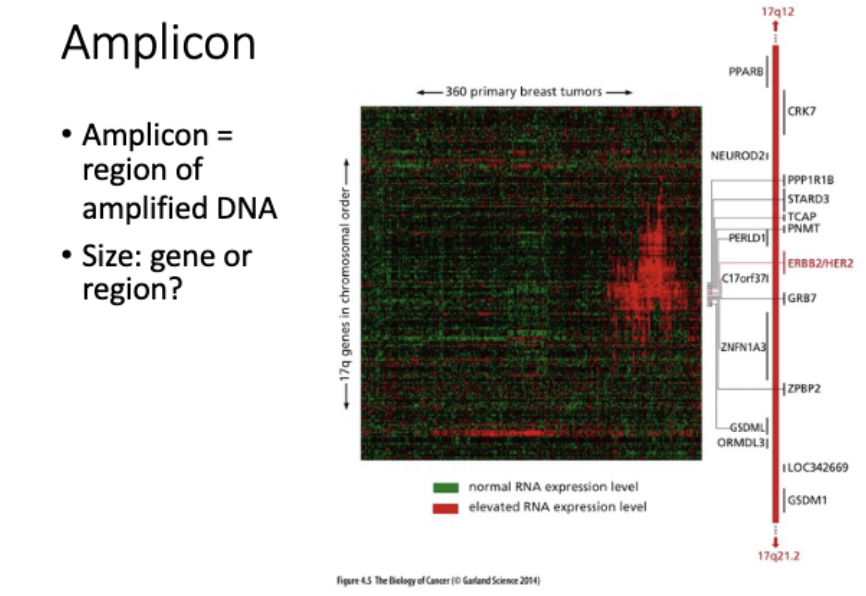
detecting amplifications - how
direct
nanopore - chr changes (LRS)
short read seq
fish
rna seq
array cgh - imbalances
indirect
western blot
rna seq- dont involve amplification
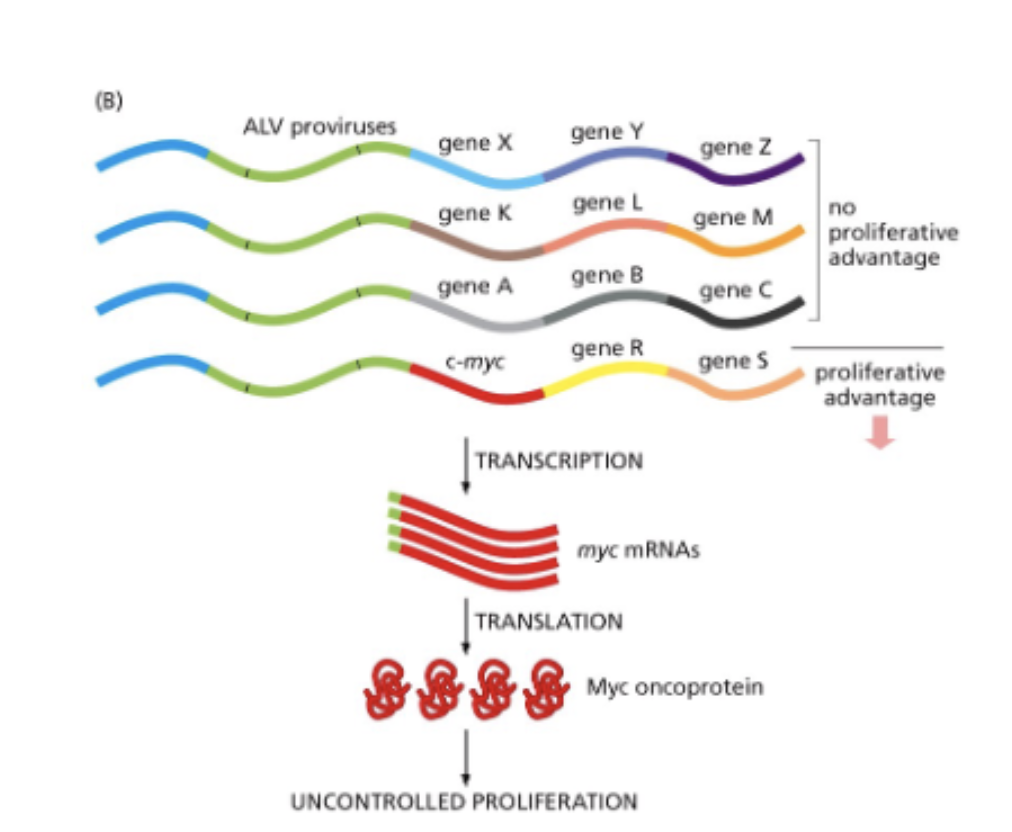
altered expression - how
viral integration (random)
promoter fustion
loss of miRNA binding sire from a transcript - loss of reg of that transcript
miRNA - genome encoded
ds miRNA processed → ss miRNA → binds mRNA → degradation, chromatin changes
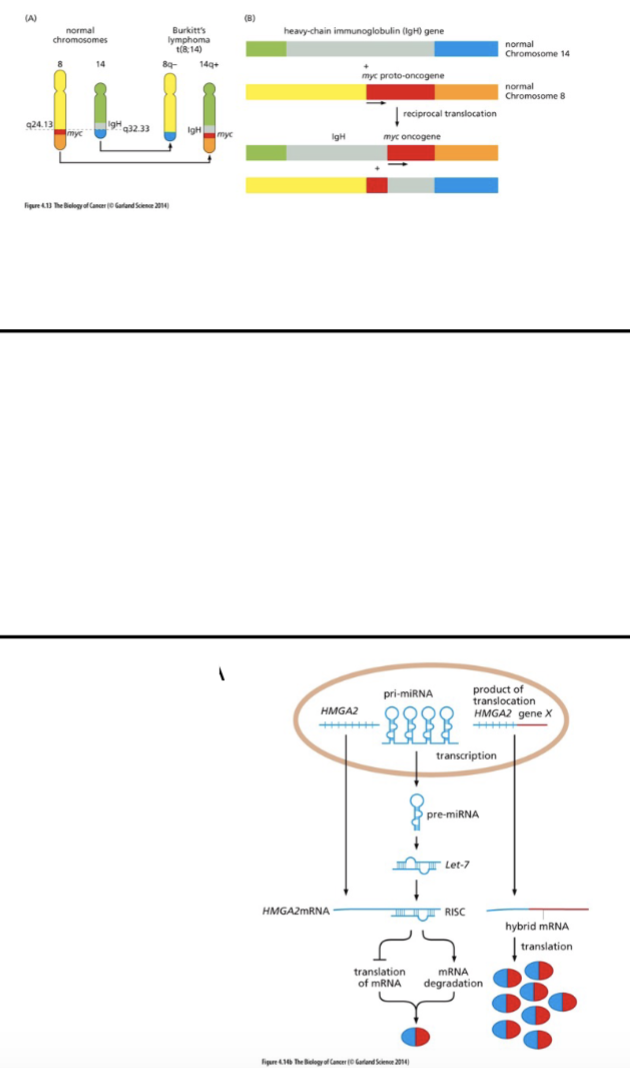
How to detect altered expression?
western blot
RNA seq
RT pcr
^^^^ not looking at DNA
northern blot
microarrays
L#16: Increased growth signaling
Explain with examples how altered function can create an oncogene and diagram how it may come about.
altered gene function
novel hybrid protein - BcrAbl (kinase)
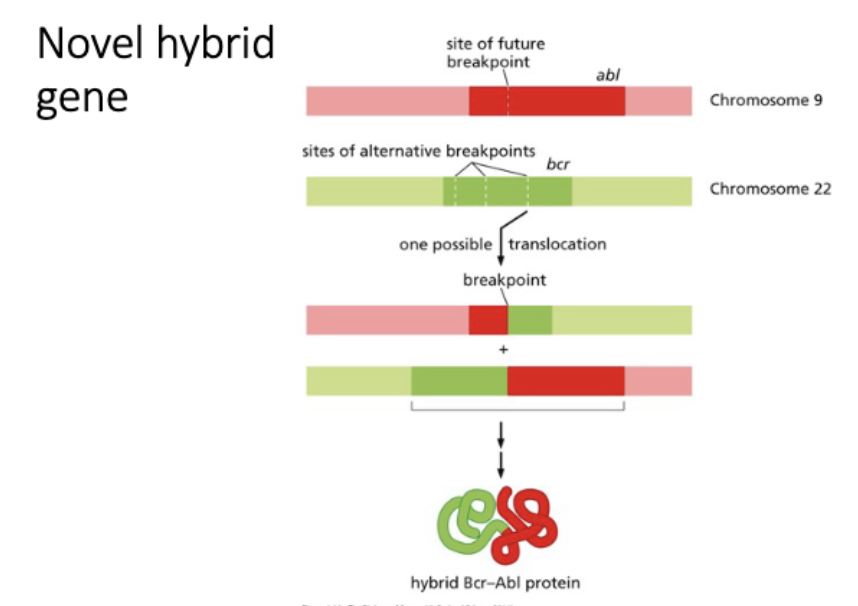
How could you test whether a novel hybrid gene is driving tumor?
TEST
sufficient - express novel hybrid → cancer
necessary - down regulate novel hybrid (RNAi, CRISPR) in tumor cells
Deregulated activity eg. Ligand-independent
signaling
truncation = growth without signal
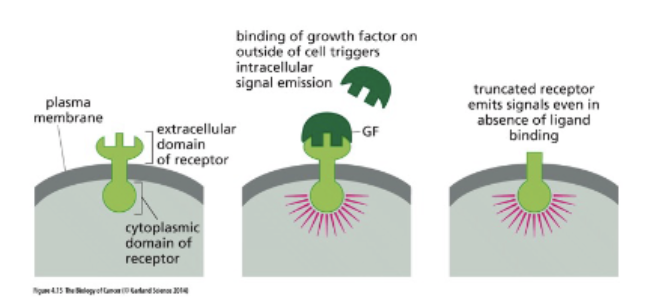
L#16: Increased growth signaling
Explain with examples how dysfunction of signaling pathways may contribute to cancer.
Explain how deregulated receptor function contributes to cancer.
Explain with examples how monomeric G proteins are activated and inactivated and how they function in signaling pathways.
Explain how dysregulation of cell cycle effectors contributes to cancer.
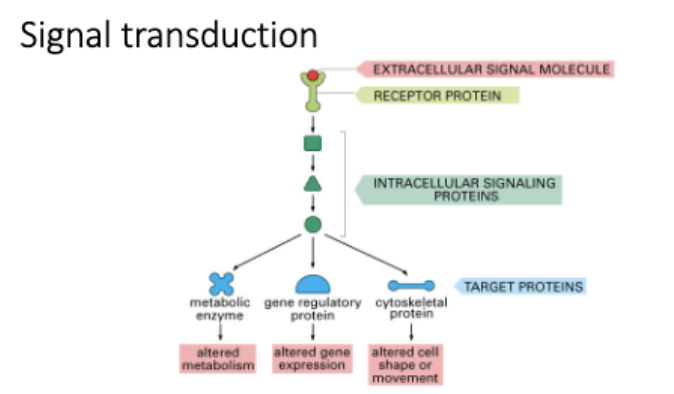
Explain how deregulated receptor function contributes to cancer.
What types of genes are proto-oncogenes?
function in signal tranduction
Ras, myc
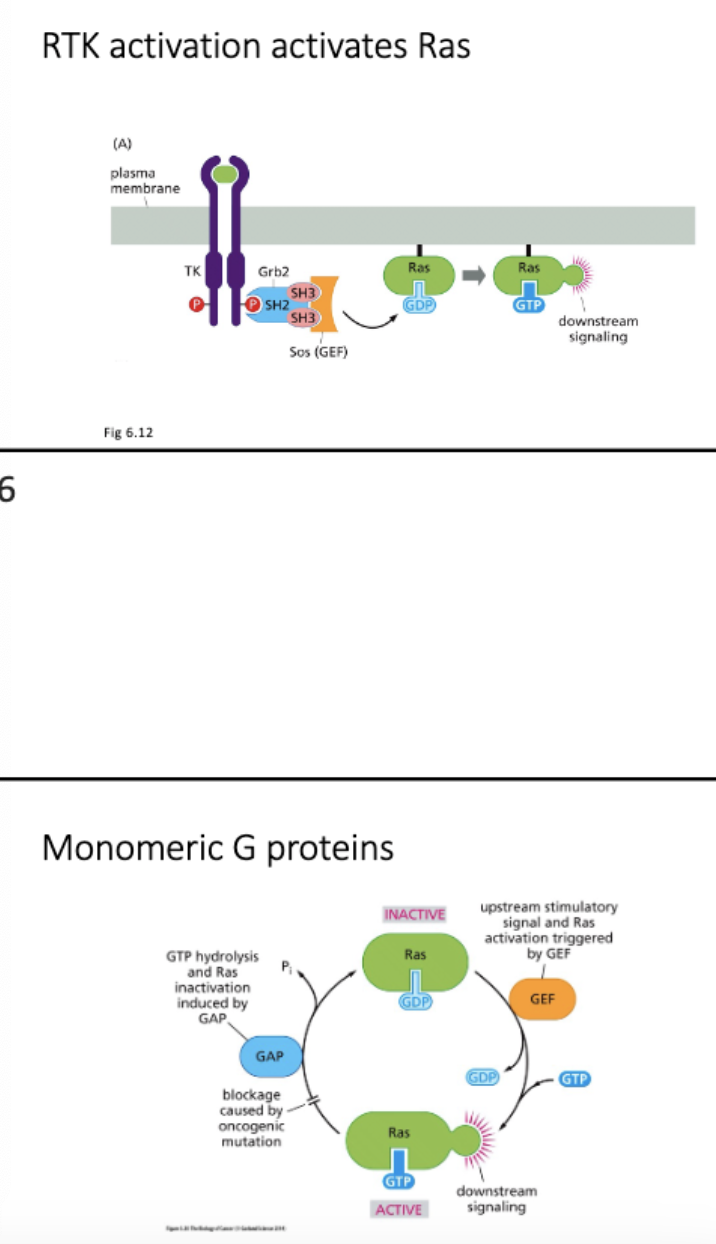
Explain with examples how monomeric G proteins are activated and inactivated and how they function in signaling pathways.
Receptor tyrosine kinase - monomers → ligand binding → dimerize → P eachother
RTK →receive prolif singal → pass to Ras → G proteins, phosphinstol
How are signals conveyed downstream of RTKs
Activate:
monomeric G protein signaling eg ras
Phosphoinositol signaling eg PI3K
Ras
L#16: Increased growth signaling
Use knowledge of signaling pathways to predict the consequence of dysfunction of particular components.
Oncogene addiction
Is the continued action of the oncogene required to sustain the cancer phenotype once a tumor is formed?
once oncogene arises, is it required for initiation or maitance?
L#17 Paper 5
Chin et al 1999
L#18: apoptosis/programmed cell death
Diagram techniques used to detect apoptosis and interpret data stemming from their use.
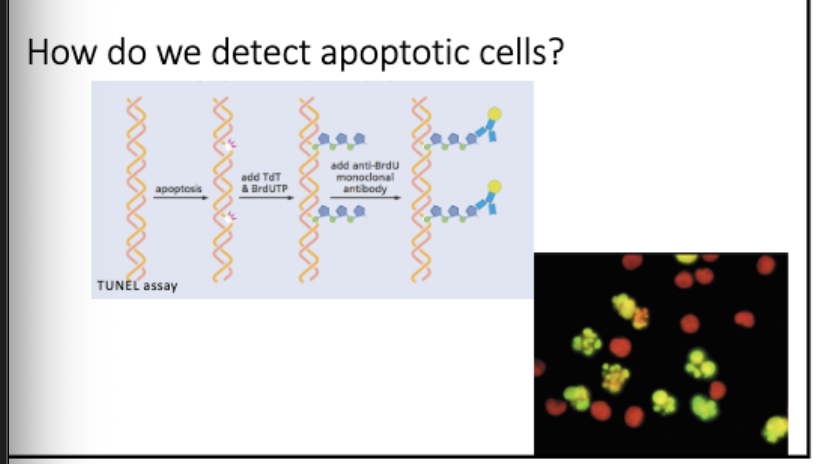
tunel assay - detects breaks during apoptosis
Tdt - terminal deoxynucleotide transferase, adds onto end
Brd UTP - nucelotide analog, recognized by Ab
add Rb to Brdu, see staining = undergoing apoptosis
apoptosis

L#18: apoptosis/programmed cell death
Describe/diagram apoptotic pathways. Predict consequences of mutations.
BCL2 - regulator of apoptosis, pro survival = ↑ tumor

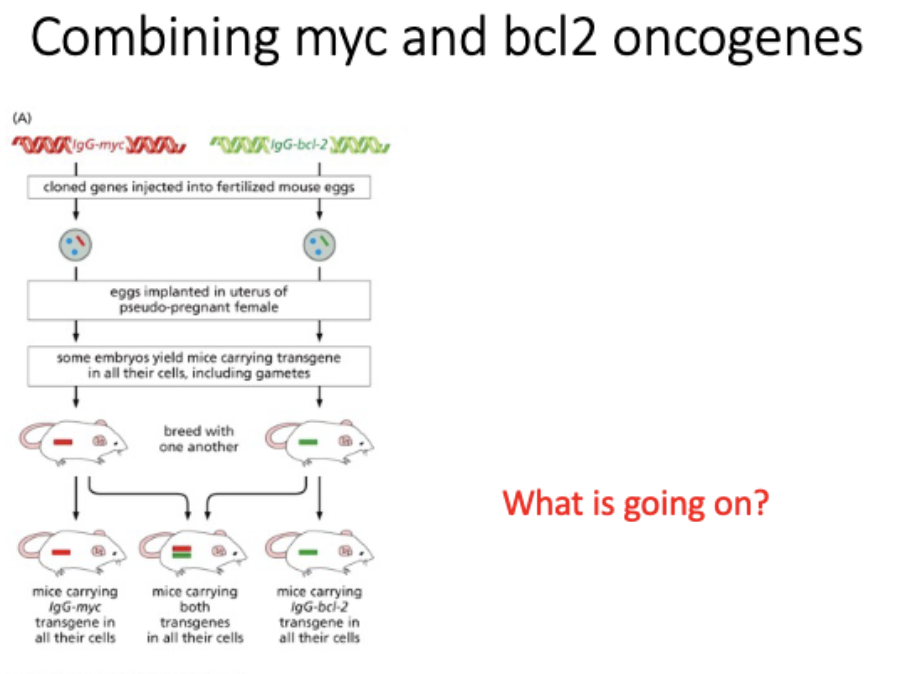

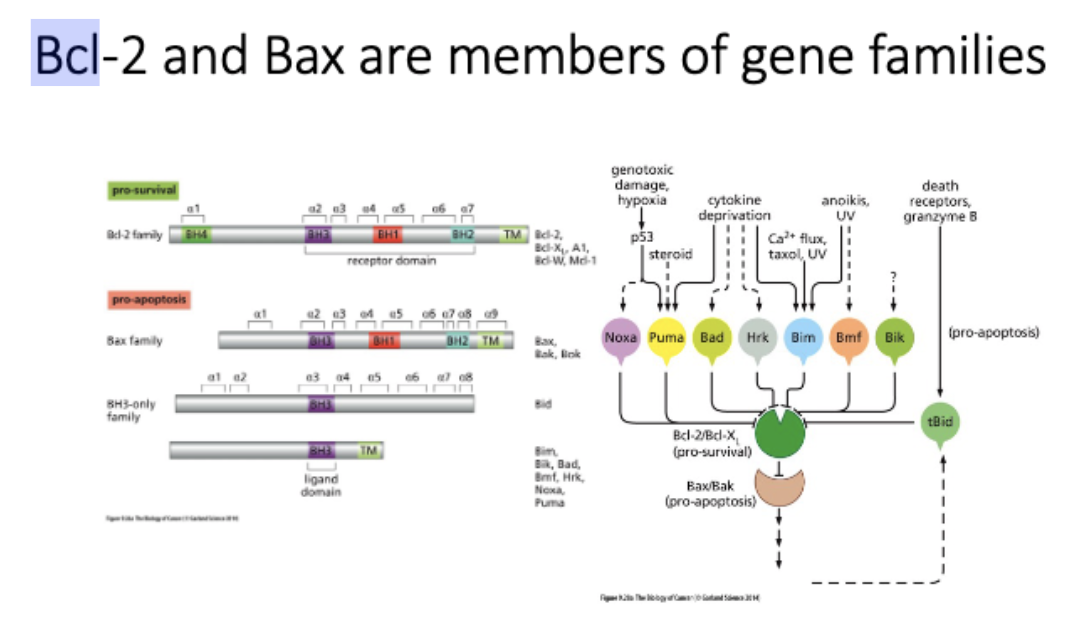
Bcl-2 and Bax are members of gene families
The balance of bcl2 and bax leads to ______ decision
life or death

L#18: apoptosis/programmed cell death
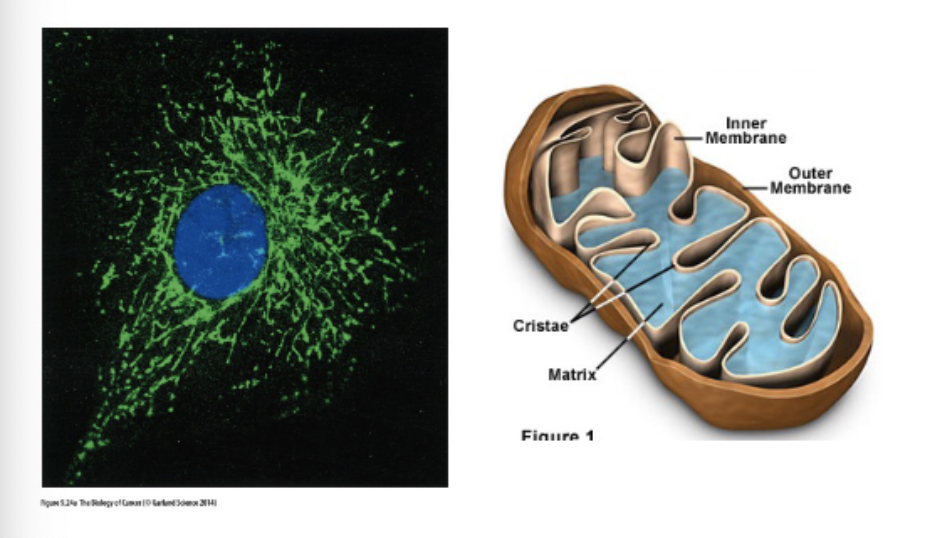
Describe the role of the mitochondria in apoptosis.
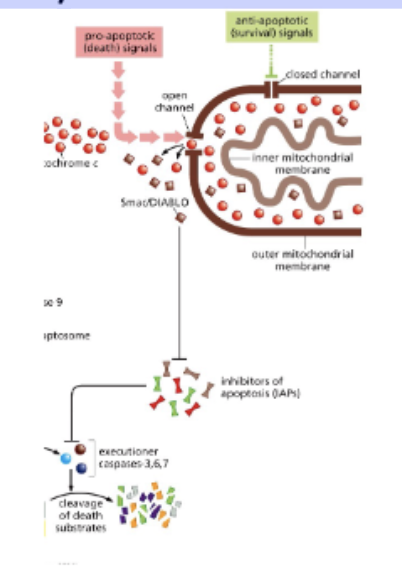
mitochondria – the home of apoptosis
anti- and pro- apoptotic signals regulate release of cytochrome c from the mitochondria
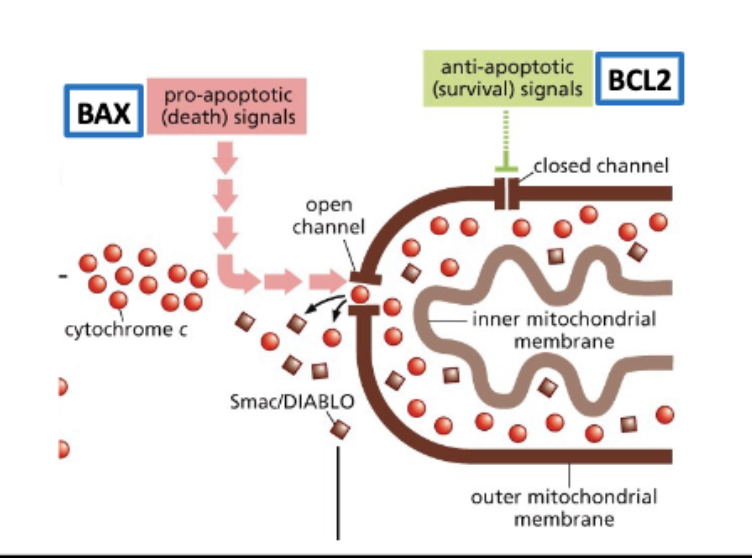
Cytochrome c release triggers apoptosis
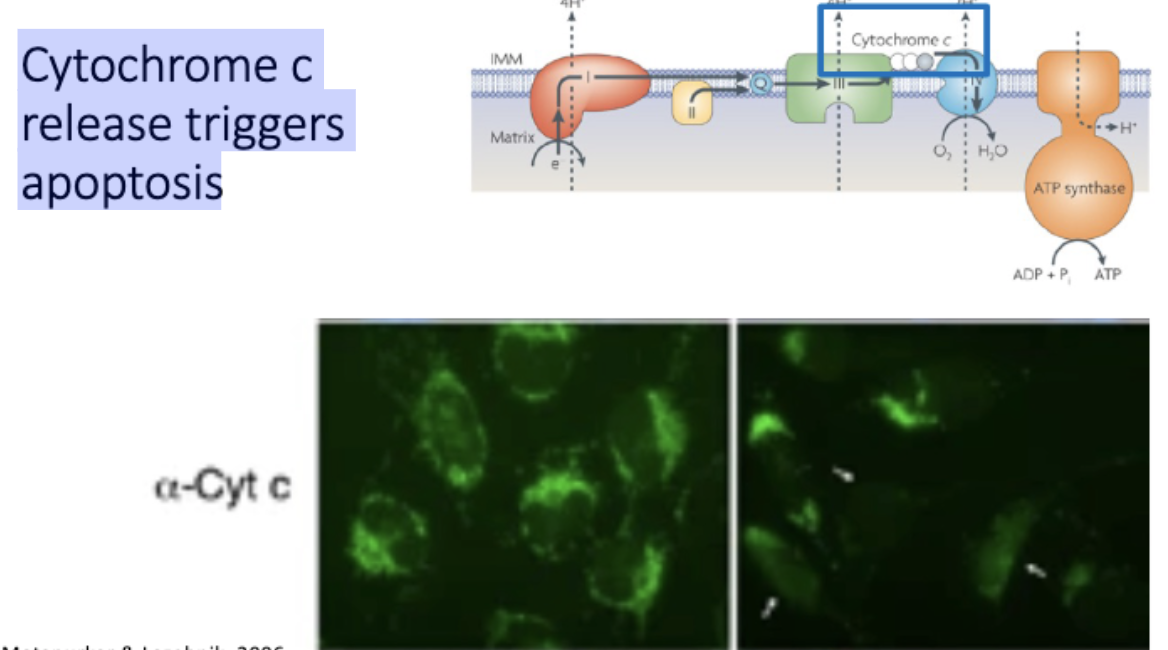
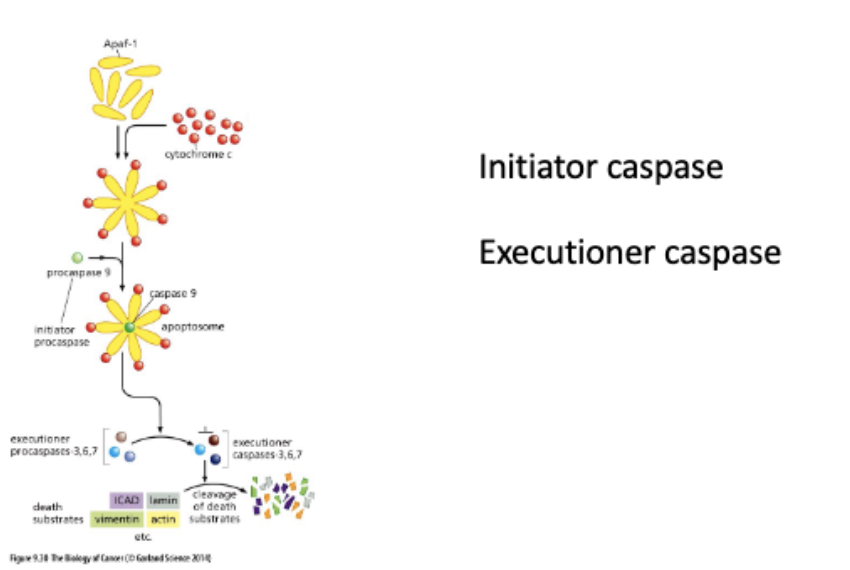
Cytochrome c triggers caspase activation
procaspase 9 → (cut) → caspace 9
proproteins - translated in inactive form
apoptosome binds + cuts other procaspaces
activates ICAD - fragments DNA
Smac/DIABLO are also released from the mt
INHIBITORS of apoptosis
L#18: apoptosis/programmed cell death
Compare and contrast intrinsic and extrinsic mechanisms of apoptosis
Where do the death ligands come from?
intrinsic -Cells can secrete the ligand to induce their own death
extrinsic - immune cells secrete death ligand

L#18: apoptosis/programmed cell death
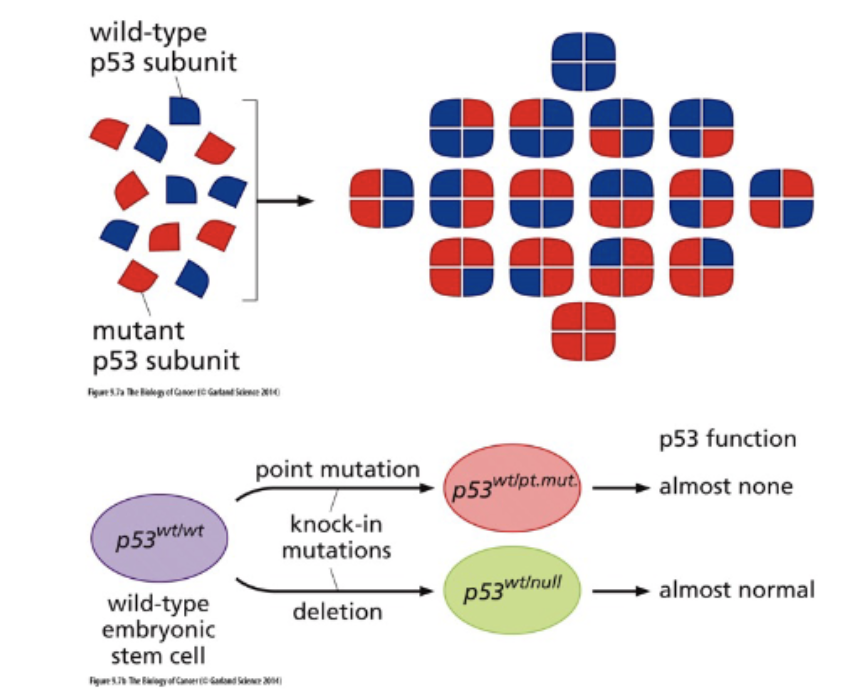
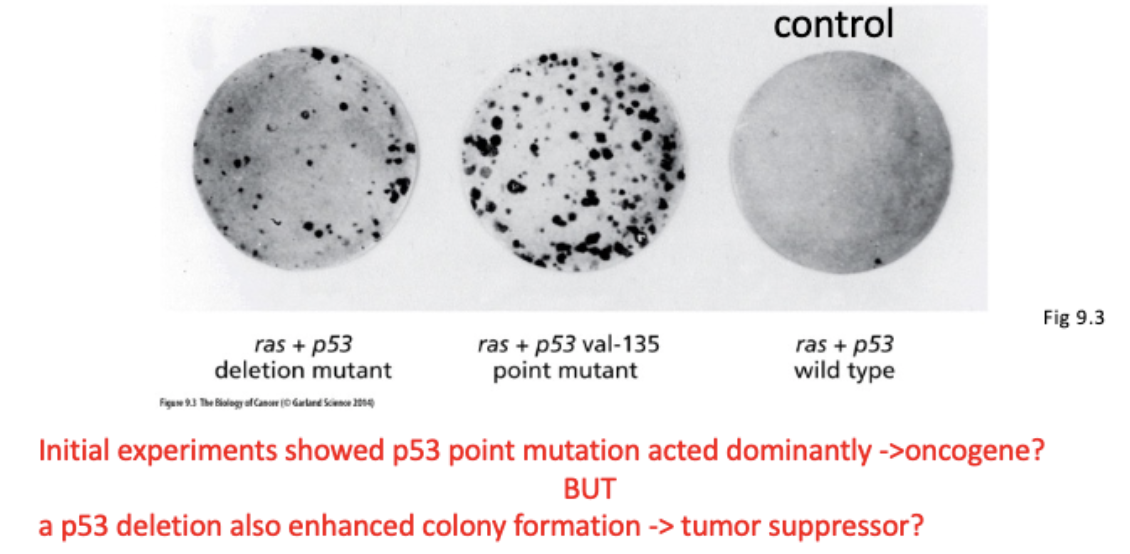
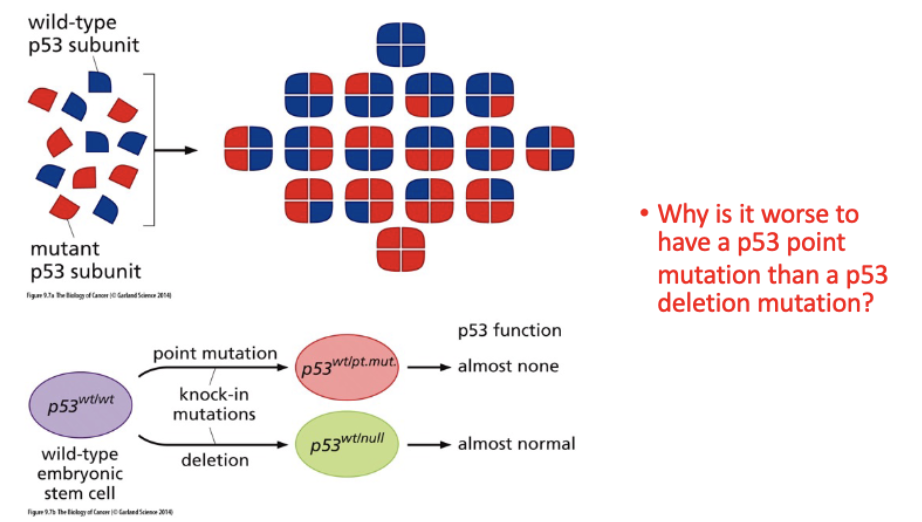
Explain how we know that many p53 mutations are dominant negative.
most frequently mutated gene in cancer
need all 4 subunits to function
Is p53 a tumor suppressor or an oncogene?
L#18: apoptosis/programmed cell death
Describe the role of p53 and how p53 mutations lead to impaired apoptosis and cancer.
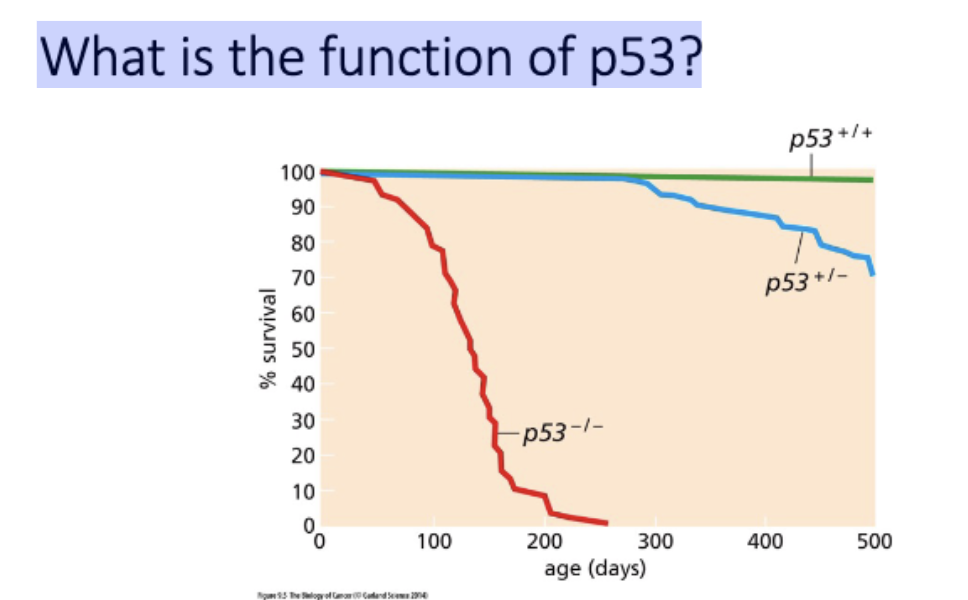
p53 - not required for development, required for longevity
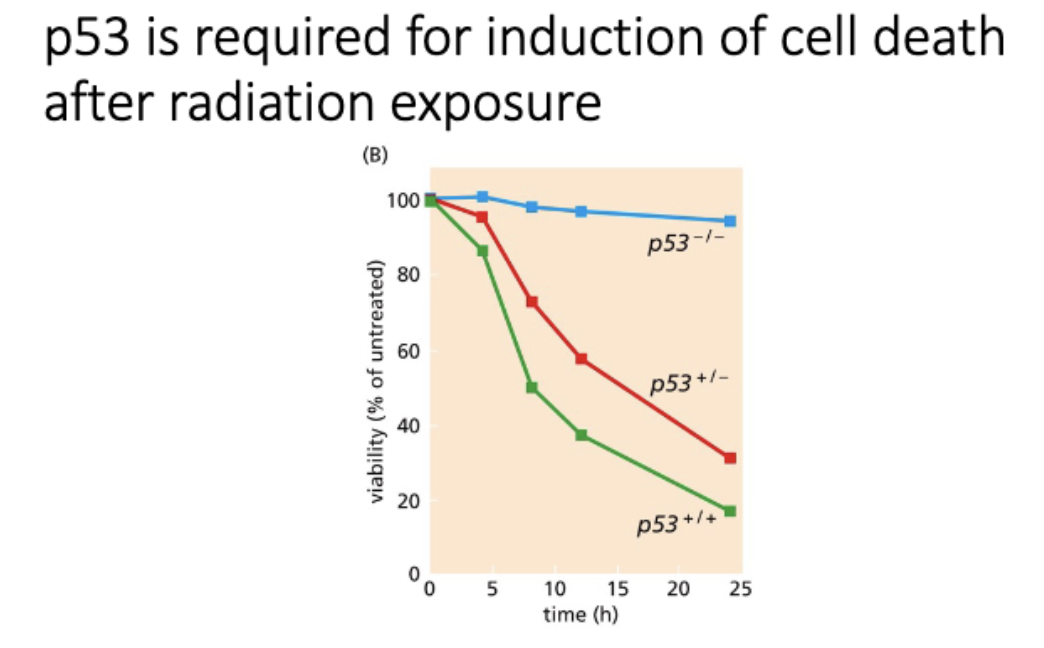
p53 required for apop after exposed to radiation
Why is it worse to have a p53 point mutation than a p53 deletion mutation?
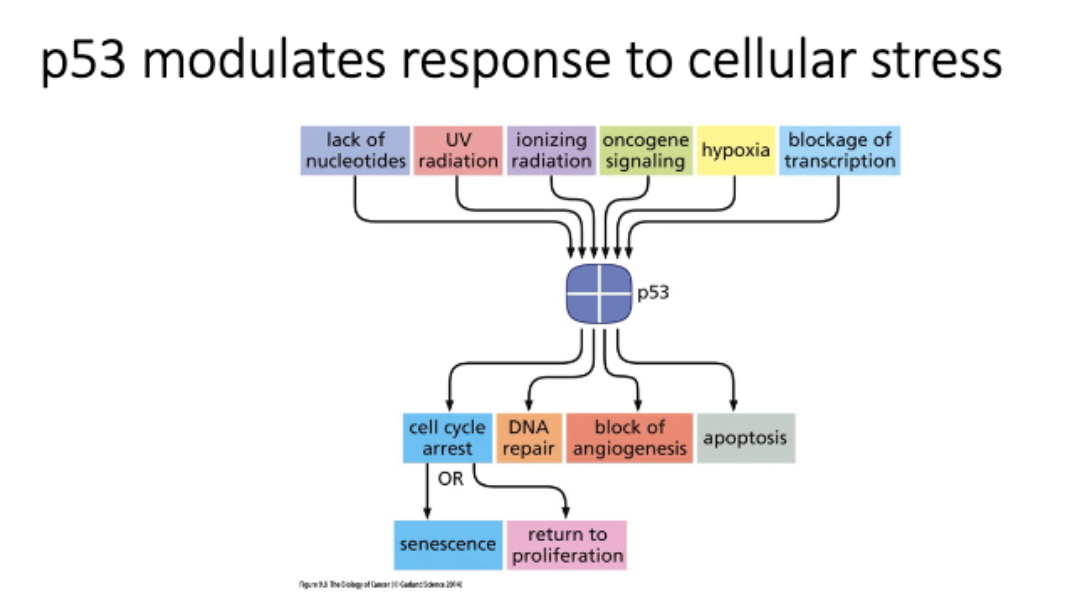
What is the function of p53?
P53 is a transcription factor
p53 modulates response to cellular stress
p53 is required for induction of cell death after radiation exposure
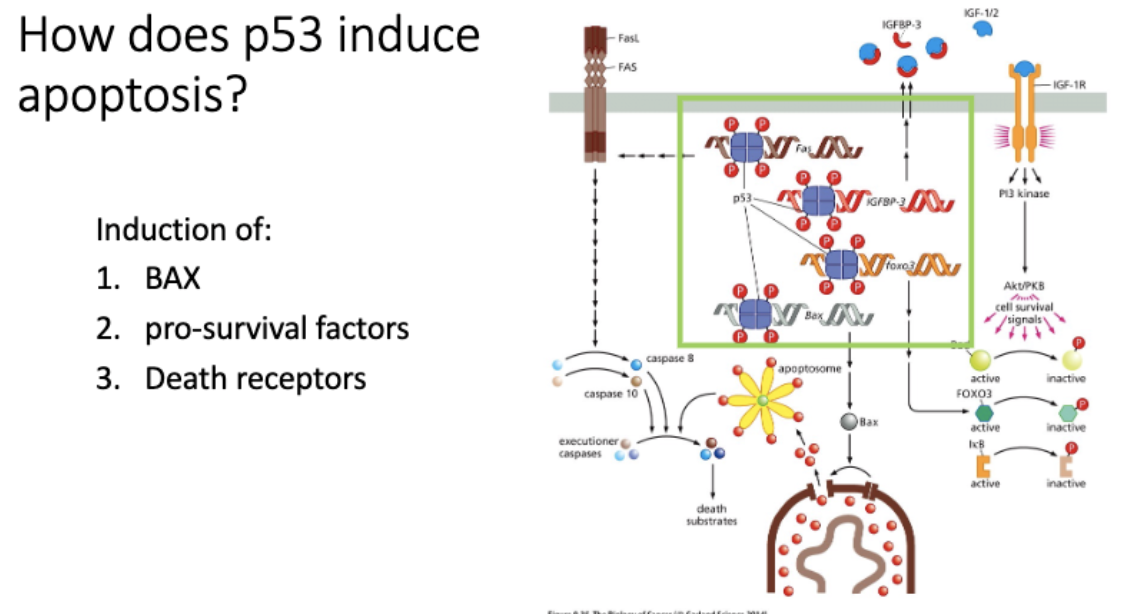
How does p53 induce apoptosis?
Bax - opens gate to mitochondria, lets out cytochrome c - induces apoptosis, apoptosome, inhibitor/executioner
FAS - cell surface receptor, receives death ligands
↑ # receptor = ↑ sensitivity to apop
* inhibits pro-survival factors
induce pro - apoptosis
L#18: apoptosis/programmed cell death
Describe the regulation of p53 and use this information to hypothesize how perturbations might affect a cell
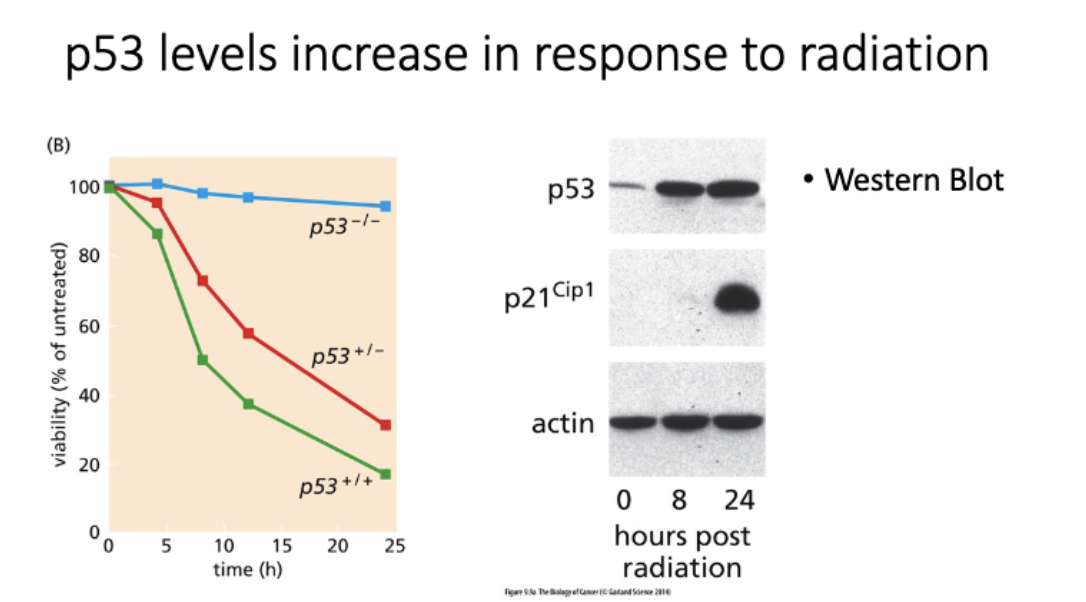
p53 levels increase in response to radiation
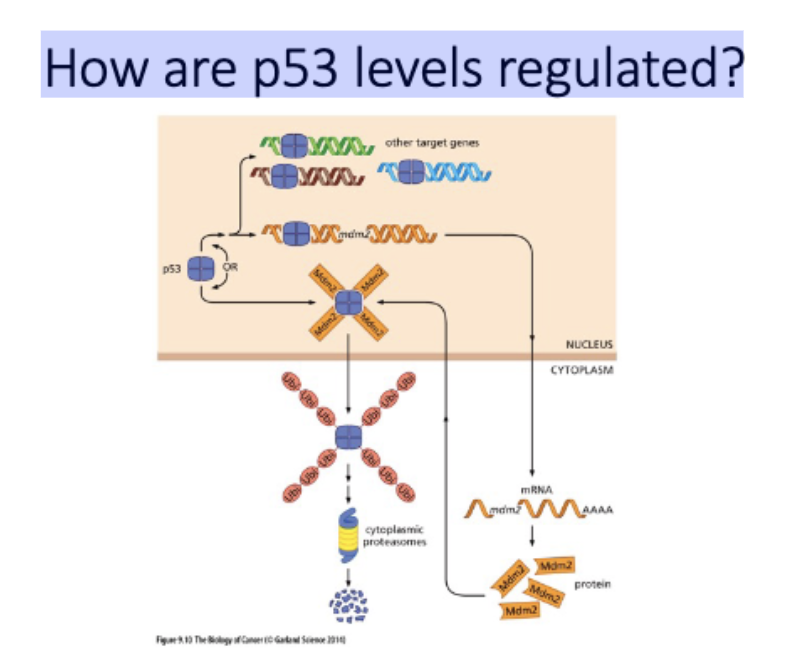
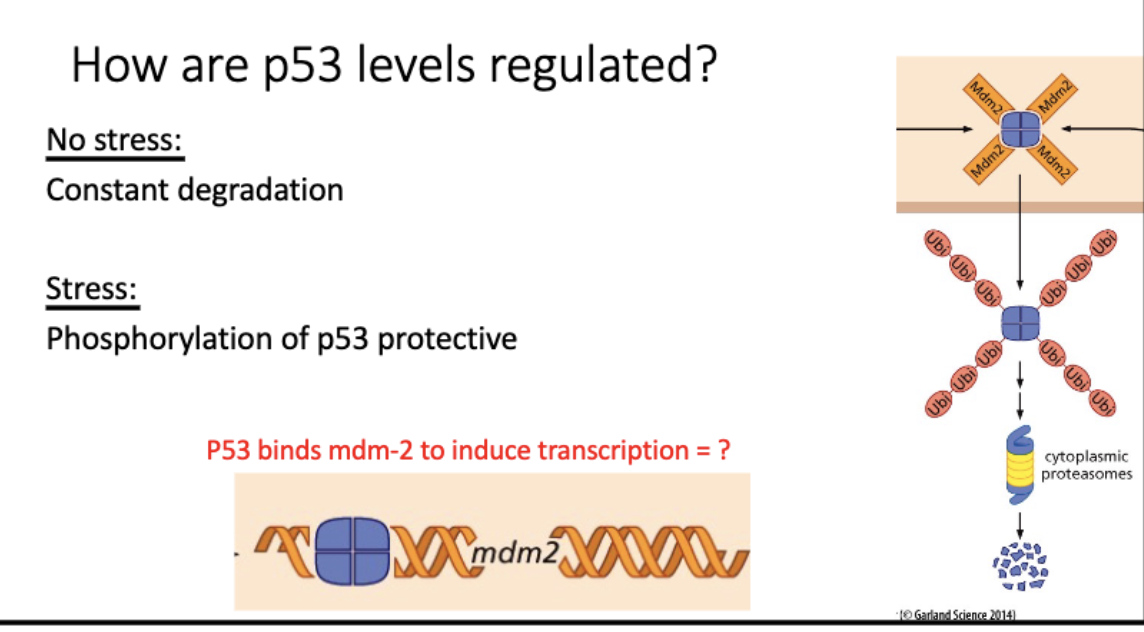
How are p53 levels regulated?
no stress - constantly made + degraded
mdm2 (e3 upiq ligase) →→ proteosome
= low p53 levels
stress - p53(P)
cant be degraded, blocks mdm2 binding
= high p53 levels
promote apop, repair, etc
p53+mdm2 promoter = transcription of mdm2 and more degradation
L#19: Why don’t elephants get cancer
C: Elephants can evade cancer through enhanced TP53 signaling.
2 models
P53 - tetramer Found at low levels in cell that is healthy growing, Levels regulated at protein level
Normal cell - p53 bound by mdm2 (e3 ubiquitin ligase, send for degradation), constantly making p53 and tagging it for degredation
DNA Damage - p53 gets phosphorylated - no longer binds to mdm, stabilized in cell
Decoy
Some of mdm binding to retrogene, leaving more p53 available under normal circumstances
Guardian
Retrogene binding to tp53 , prevents it from being degraded by mdm2
Found by immunoprecipitation that rtg bound to tp53 and NOT mgm2
Computational modeling - dimers more stable