NURS2001 - Wk 2 lec Pharmacokinetics
1/27
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
28 Terms

Pharmacokinetics
What the body does to the drug (does & conc of drug in the blood)
Pharmacodynamics
What the drug does to the body (pharmacological effect)
Drug action & Disposition of Pharmacokinetics
ADMe =
Distribution (bound or unbound to plasma proteins around body), Absorption (release of drug), Metabolism, elimination (Kidney —> Urine, or Liver —> Bile)
Methods of medication absorption and routes of its administration
Journey of drug molecule into the plasma (blood) through to circulation
Routes:
Oral
Sublingual
Rectal
Inhalation
Injection (eg. Subcut, IV, intramuscular)
Oral Administration Absorption - where does it occur?
Occurs through GIT
Stomach
Mucous barriers on stomach lining reduce absorption, + low pH (acidic environment favours weak acid)
Drugs can be protected from the acidic pH with an enteric protein coat surrounding the drug (should the drug be weak in low pH)
Small Intestine
Large SA (villi & microvilli), No mucous barrier, fastest Gi site absorption for drugs
Large Intestine
Less villi, Slower absorption compared to SI
Factors affecting oral absorption
Gastric emptying
Rapid emptying (before meal) gives rapid absorption into SI, should you take a drug with a faster onset of action
Slow emptying (with food) for slower absorption to give the drug more time to be absorbed
GI motility
Increased motility (eg. diarrhoea) causes decreased absorption
Bioavailability (F)
Fraction of dose that is absorbed into the systemic circulation (0%-100%)
Importance of Bioavailability
To determine dosages for non-IV routes of administration (since IV administrated drugs = 100%, F = 1)
Bioequivalence
Two products that have the same rates of absorption and bioavailability
Important to be able to tell whether different brands of the same drug can be interchanged
First pass hepatic elimination
When a drug is metabolised in the liver before it reaches the systemic circulation, reducing Bioavailability
Why some drugs are not given orally etc. due to high first pass effect
Distribution
How the drug flows throughout the body once entered the systemic circulation
What affects distribution?
Drug characteristics:
Permeability between plasma membranes, size (unbound drug molecules can transport into different compartments), pKa, weight
Physiological characteristics:
Blood flow, Cardiac output
Plasma protein binding
Unbound drug molecules bind to proteins to form drug-protein complexes
Allows for increased release of unbound drug
Bound drugs act as a reserve, ready to release (as binding is a reversible reaction) thus allowing for a slower but extended release of active unbound drug molecules
Volume of distribution (Vd)
Volume of fluid required to contain the total amount of drug in the body at the same concentration as plasma that is present
Determined by:
fu
Tissue binding
It is a ratio between drug concentration and the amount of dose in the body.
The drug dose required to uniformly distribute throughout the body, hence high and low Vd is a significant distinguisher
Vd significance
Significance of Vd
Drugs w/ small Vd
Shorter half life
Accumulate less in the body
Safer for use during pregnancy and breastfeeding
Drugs w/ large Vd
Longer half lives
Accumulate more in the body
Requires loading dose (large initial dose for drug to reach therapeutic effect levels in the body
Large vs low Vd
Large = More likely to be found in the fats/tissues of the body
Low = More likely to be found to the blood circulation (confined)
Clearance (CL)
How efficient drug elimination is
Which allows for the determination of the appropriate dose
Half life
How long a drug stays in the body
How long it takes the plasma concentration of the drug to reach 50%
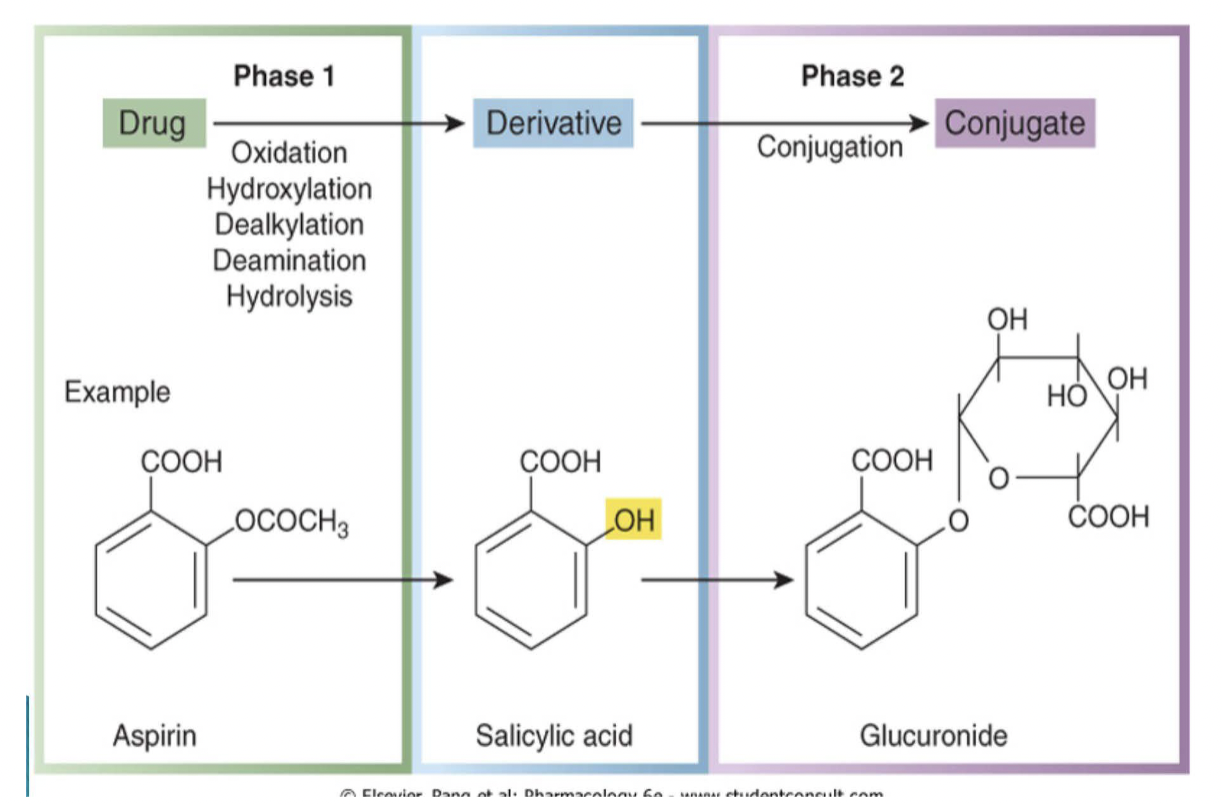
Two phases of drug metabolism
Phase 1: OHDDH
Oxidation
Hydroxylation
Dealkylation
Deamination
Hydrolysis
Phase 2:
Conjugation
Routes of elimination
Metabolism
Excretion via renal and biliary
Exhalation
Cytocrome-P450 main enzymes
CYP3A4
CYP2C9
Enzyme forms
Substrate = Metabolised by or compete for metabolic (enzyme) sites
Inhibitor = Competes and blocks metabolic sites (INHIBITS)
Inducer = Increases metabolic activity by increasing amount of enzymes (INDUCING)
Differences between the effects of inhibitors and inducers
Inducers = Enhance CYPs effect + Speed up metabolism of drug
Inhibitors = Reduce CYPs effects + Slow down metabolism of drug
Inducers examples
Cruciferous vegetables
Phenytoin
Inhibitors examples
Clarithromycin
Erythromycin
Pharmacokinetic Parameters
Clearance (CL)
Efficiency of drug elimination
Volume of Distribution (Vd)
Extend of distribution in the body eg. fat, tissues, systemic circulation
DETERMINES THE LOADING DOSE
Bioavailability
Fraction of a dose absorbed into systemic circulation