Anticancer Agents
1/84
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
85 Terms
Cell Cycle
process by which cell multiplies
in healthy cell, has many checkpoints
cell cycle is promoted by:
Cyclins: molecules that activate CDKs, ex: Cyclin A, Cyclin B, Cyclin D, Cyclin E
Cyclin Dependent Kinases: receptors that are activated by Cyclins (ex: CDK-1, CDK-2, etc.)
Cyclin D is particularly important (G1 to S Phase?)
Cancer occurs when problems occur with the regulatory process
note: kinase means phosphorylation
Genetic vs Epigenetic
Genetic:
based on gene sequence
Epigenetic:
not dictated on gene sequence, but rather genes turning “on or off”
Carcinogenesis
Starts as Normal Cells
Exposed to Carcinogens
Types of Carcinogens:
Initiators: UV Light, charred food (benzo-α-pyrenes), carcinogenic chemicals, etc.
Promoters/Potentiators: benzo-α-pyrenes (result of incomplete combustion of organic material, aka char),
need exposure to an initiator, then continuous exposure to potentiator to develop cancer
Process: Normal → Initiation → Promotion → Progression → Tumor → Metastasis
body can repair damage, but only so much
Note: P53 - aberration in P53 gene is associated with 80-90% of colon cancer
Detoxification
Phase I and Phase II of biotransformation
aka when drugs are transformed in the body
especially important: Phase II reactions (glucuronidation, conjugation reactions, etc.)
for this context, activates in response to initiators
transforms them into entities safe for excretion
Phase I (CYP) enzymes go down, Phase II enzymes go up in response to initiators
due to Phase I possibly activating instead, like prodrug (ex: benzo-α-pyrenes)
Progression and Proliferation
Progression:
cell responds to damage by triggering apoptosis and cell cycle arrest
inflammation occurs
eventually, increased mutated cell count promotes angiogenesis to gain nutrient supply
note: vessels formed this way are less stable and structurally sound
metalloproteinases are activated to break down ECF and allow easier angiogenesis
Invasion and Metastasis
metalloproteinases are activated to break down ECF and allow easier angiogenesis
eventually, with enough progression, cancer cells invade
move from site of initial tumor to other sites (called metastasis)
Metastasis is usually at around Stage IV Cancer
Tumors that do not metastasize are benign tumors, are generally encapsulated
metastatic tumors are called malignant, generally not encapsulated
metastatic cells begin harming organs and body
ex: finding colon cells in lungs means metastasis has occurred
site for invasion are generally highly vascularized tissues
Proto-Oncogenes and Tumor Suppressor Genes
Mutations in either of these genes cause cancer
Proto-Oncogenes:
when expressed, generally promote cell division
when mutated, becomes oncogene
mutations promote the gene, leading to unregulated cell division
ex: genes for Growth Factors, Tyrosine-Kinases (GF Receptors are Tyrosine Kinases), Transcription Factors (initiate RNA Polymerase action)
Tumor Suppressor Genes:
when expressed, generally inhibit cell division
mutations silence the gene, leading to unregulated cell division
ex: genes for P53, Tumor Growth Factor β
EGFR Signaling Pathway
or endothelial growth factor receptor signaling
downstream effects lead to activation of endothelial growth factor
receptors found in capillaries; when activated leads to more capillaries forming
abnormally activated leads to angiogenesis
Note: silencing of Tumor Growth Factor β and P53 can lead to this?
RAS Protein
Pathway:
growth factor acts on tyrosine-kinase GF receptor on cell surface
activation of receptor leads to either cell growth or production of more receptors
RAS protein is connected to receptor by bridging protein; GF Receptor Kinases phosphorylate bridging protein
phosphorylation activates RAS; bound GDP becomes GTP, detaches from bridging protein
RAS activation leads to activation of other proteins down the line, leading eventually to cell division
RAS is a regulatory protein
mutation (ex: perpetual activation) leads to uncontrolled cellular division
Apoptosis
programmed cell death
does not cause inflammation (cell remains are enclosed in membrane after death)
Caspases: proteases that are instrumental to apoptosis
signals that inhibit their function can limit apoptosis
limited apoptosis is a symptom of cancer
Intrinsic Pathway:
cell is damaged, ex: exposed to DNA damaging agent
cell recognizes damage and if can not repair, self-destructs
involves p53
Extrinsic Pathway:
signals from the body send a signal to the specific cells at a specific time to die
supposed to happen, not due to damage
caused by extracellular agents
Telomeres and Telomerase
Telomere:
nucleic acid chain at the end of DNA
protects DNA
shortens every replication
if too short, eventually reaches senescence and cell death
Senescence: state where cells stop dividing
if telomeres do not get shorter, unregulated cell division may occur
note: longer telomerases also may prevent cells from maturing and differentiating from stem cells
Telomerase:
rebuilds or creates telomeres
inhibition is a target for anticancer agents
Angiogenesis
formation of new capillaries
used by tumors to provide nutrition to areas unreached by normal blood vessels
relies on VEGF (Vascular Endothelial Growth Factor) and VEGF receptors
tumors release growth factor
vessels will create receptors or new endothelial cells
new vessels are formed
for tumors, hasty vessel
Matrix Metalloproteinases
Extracellular Matrix (ECM) cements cell in place, improves integrity (ex: collagen)
also forms barrier preventing easy innervation of blood vessels
makes angiogenesis harder
MMP’s dissolve matrix
in cancer, promotion of MMPs weakens matrix, promotes tumor and angiogenesis
drug target: deactivation of MMPs
Mesenchymal and Parenchymal Cells in Regards to Cancer
Parenchymal Cells:
functional cells
adherent, tend to stay in one place
Mesenchymal Cells:
multipotent cells
motile, have ability to move
can survive blood flow and pressure
Metastasis relies on the ability to interconvert
cancer cells are pleomorphic; shift to motile mesenchymal cells to move to new places
then shift to adherent parenchymal cells to anchor in favorable places
Available Cancer Treatment
3000 BC to 1980: Surgical Treatment
1900: Radiotherapy
note: may develop secondary tumor elsewhere after treatment
cancer cells may simply move to different spot after treatment
1940: Chemotherapy & Hormonal Therapy
fun fact: many were inspired by nerve gas
this is why the wartime effort helped with development
2000: Targeted Therapies & Monoclonal Antibodies
enzymes and chemotherapeutic agents are conjugated to monoclonal antibodies so that drug is directed to target cancer cells
2013: Checkpoint Inhibitors & Engineered Cell Therapy
example, CAR-T Cells
Cancer Cell Drug Resistance
extremely common: efflux proteins (ex: ATP-dependent pumps)
decreased drug influx
modification of drug target proteins (ex: telomerase, etc.)
drug compartmentalization (ex: in lysosomes)
Increased drug deactivation via metabolism
Enhanced DNA Repair
Inactivation of Apoptotic pathways
etc.
Intrinsic vs Acquired Resistance
Intrinsic Resistance
cancer cell is resistant from the beginning
slow growth rate
poor drug uptake
biochemical/genetic properties of the cell
Acquired Resistance:
used to be susceptible, but then cancer cell mutated
presence of drug-sensitive and drug-resistant cells within tumor
mutation results to abnormal number/structure of protein drug targets in cells
Drug Targets: Summary
Note: cancer is a systems biology disease
even if one mechanism/target is attacked, cancer may just find another pathway to survive
systematic approach to attack multiple targets with compatible drugs is recommended
Drug Targets: Monoclonal Antibodies
also called MABs
not chemotherapeutic
lab made antibodies, designed to target a single, specific target
-zumab, -imab, -mab at the end
can inhibit:
Growth Factor
Binding of Growth Factor (direct or allosteric)
Release of Growth Factor from Cancer Cell
Drug Targets: Hormones
some cancers are hormone dependent (note: not all cancers in hormone areas are hormone dependent, do genetic test)
ex: breast cancer does not always mean estrogen dependent
some hormones are transcription factor producers/act as transcription factors
target: hormones with antihormones
attack either hormones or their receptors
note: hormone receptors tend to be intracellular (due to lipophilic ligands)
ex: estrogen in breast cancer
Drug Targets: RAS Protein
Farnesyltransferase Inhibitors (FTIs)
Farnesyltransferase is the protein that connects to RAS and associated protein
inhibition of FT leads to inhibition of RAS activity
Drug Targets: Tyrosine Kinases
GF receptors tend to be tyrosine kinases
inhibit receptors from the inside of the cell
must enter cell to work
blocks the cascade leading to RAS activation
generally look like and are competitive inhibitors of ATP (because kinase)
-inib at the end
Drug Targets: Mitosis
mitotic disruptors
selective towards mitotic spindle, which only forms in actively dividing cells
prevents cell proliferation
ex: vinca alkaloids
-blastine, -istine, etc.
Drug Targets: Telomerase
epigenetic
by targeting telomerases, prevent prolonged cell lifespan and division
Drug Targets: Histone Deacetylase
epigenetic
Histone: protein in nucleus, DNA wraps around histones to form chromatin
When histones are deacetylated, allows DNA to bind to them
DNA has negative phosphate groups, which bond to the positive deacetylated histone
inhibiting deacetylase prevents histones from acting
this unwinds/unsilences Tumor Suppressor Genes
note: promoting histone acetylase will do the same thing
note: inhibiting histone acetylase for oncogenes will also have effect
Deacetylase Inhibitors must be very specific for Tumor Suppressor Genes; Acetylase Inhibitors must be specific for Oncogenes
due to complexity, few are yet on market
Drug Targets: Topoisomerases
relieve torsion stress when DNA is unwinding
inhibition prevents DNA unwinding, and breaks DNA
effectively stops DNA duplication and mitosis
Drug Target: Antimetabolites
fake pyrimidine/pyridine bases (ex: fluorouracil)
can also be folic acid antimetabolites
prevent elongation of DNA
cell takes them up as if they are real bases
incorrect structure prevents further DNA addition
Base Analogues are prodrugs, are triphosphorylated by cell itself
if pre-phosphorylated, too polar to enter through membrane or transporters
while repair mechanism will fix, it will eventually be overwhelmed
Note:
Thymidine in DNA
Uracil in RNA
Drug Targets: Chain Cutters
cross-link with DNA
form ROS (Reactive Oxygen Species) near DNA which attack and damage DNA
eventually cuts DNA chains
Drug Targets: Nucleic Acids
target DNA, RNA, or their production
Intercalating Agent:
insert between DNA base pairs and cause damage and error
Non-Intercalating Agent:
can inhibit topoisomerases
Chain Cutters:
form ROS and attack chains
Alkylating Agents:
old school among chemotherapeutic agents
form cross-links on the strands themselves and change structure
prevents recognition by polymerase; either is repaired or process halts
Antimetabolites:
folic acid, or actual nucleic acid bases
Antisense Therapy:
uses antisense nucleotides to target specific mRNA to turn on/off specific genes
angy dia

Topoisomerases
relieve torsion stress in DNA unwinding
Types:
Topoisomerase I:
only capable of single strand breaks
must cut DNA strand to remove supercoiling
Topoisomerase II:
capable of double strand breaks
Note: suicide substrates (die with receptor) can kill topoisomerases (topoisomerase poisons)
when topoisomerase is damaged; will be disposed of by proteasome
Note: some drugs inhibit by attaching to both DNA and topoisomerase, while some just attach to the DNA
Topoisomerase Inhibitors (Intercalating): Anthracyclines
Intercalator
ex: doxorubicin, daunorubicin
planar, flat structure of anthraquinone part allows to fit in between bases in DNA chain
Nitrogen in DNA bases attacks N-C Carbon on sugar-amine part; alkylates the DNA
prefers Cytosine and Guanine due to hydrogen bonding between bases and drug, and van der Wals
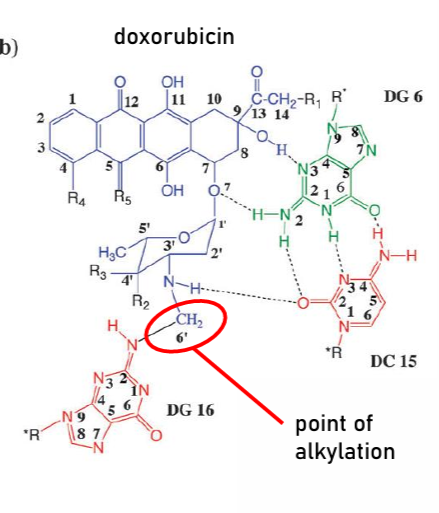
technically also inhibits DNA Polymerase
also generates ROS to damage DNA
Side Effects:
cardiotoxic
has high affinity for cardiac tissue
sugar-amine + planar = hydrophilic head, hydrophobic tail emulation
incorporates into cell membrane
increases drug permeability; general membrane disruption
also ROS generation can cause toxicity
ROS generation due to anthraquinone nature (can form quinone radicals)
Topoisomerase Inhibitors (Intercalating): Mitoxantrone
intercalating agent
stabilizes near DNA due to hydrogen bonding
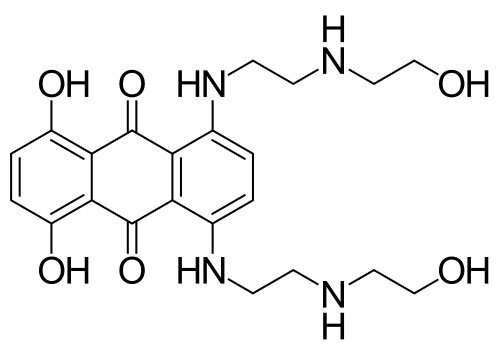
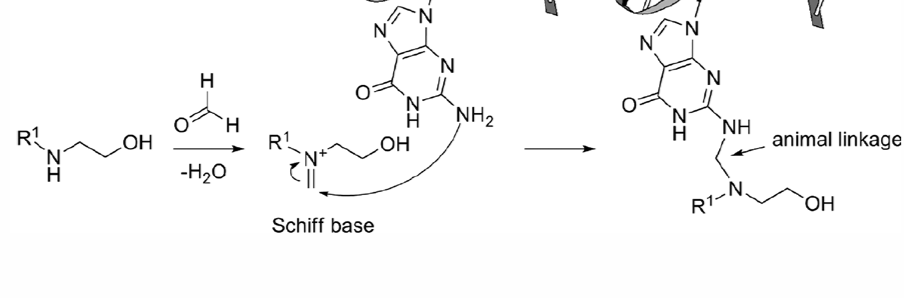
Mechanism:
forms Schiff Base (Imine) with any nearby carbonyl
Imine is electrophile, Nitrogen in DNA attacks carbon attached imine
forms alkylation
favors guanine residue
Topoisomerase Inhibitors (Intercalating): Dactinomycin
peptide polyketide
triplanar, intercalating agent
attaches to minor groove of DNA
positively charged amino form is predominant in solution
forms ionic bond with DNA negative phosphate backbone
inhibits Topoisomerase II
favors C-G Pairs (due to Hydrogen Bonding)
Topoisomerase Inhibitors (Non-Intercalating): Etoposide
intercalating agent
from natural product
can bind with either topoisomerase, DNA, or both
Topoisomerase Inhibitors (Non-Intercalating): Camptothecins
can intercalate, but not generally known for that
via van-der Wals forces (Rings A & B), and hydrogen bonds
can bind to Topoisomerase I and DNA at the same time
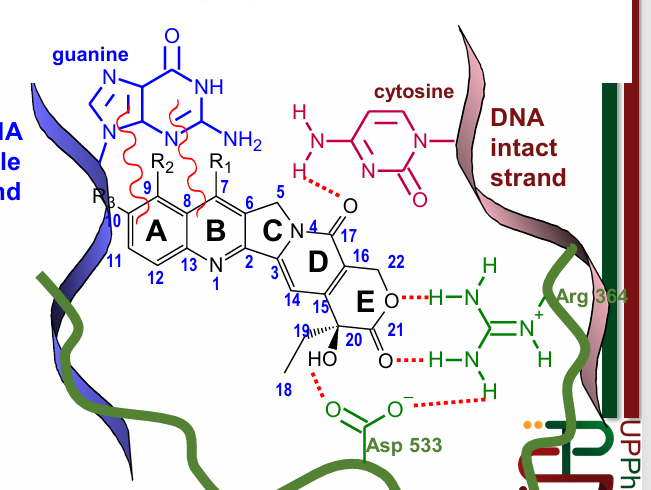
cell cycle nonspecific: toxic even to non-replicating cells
ex: topotecan (ovarian cancer), irinotecan
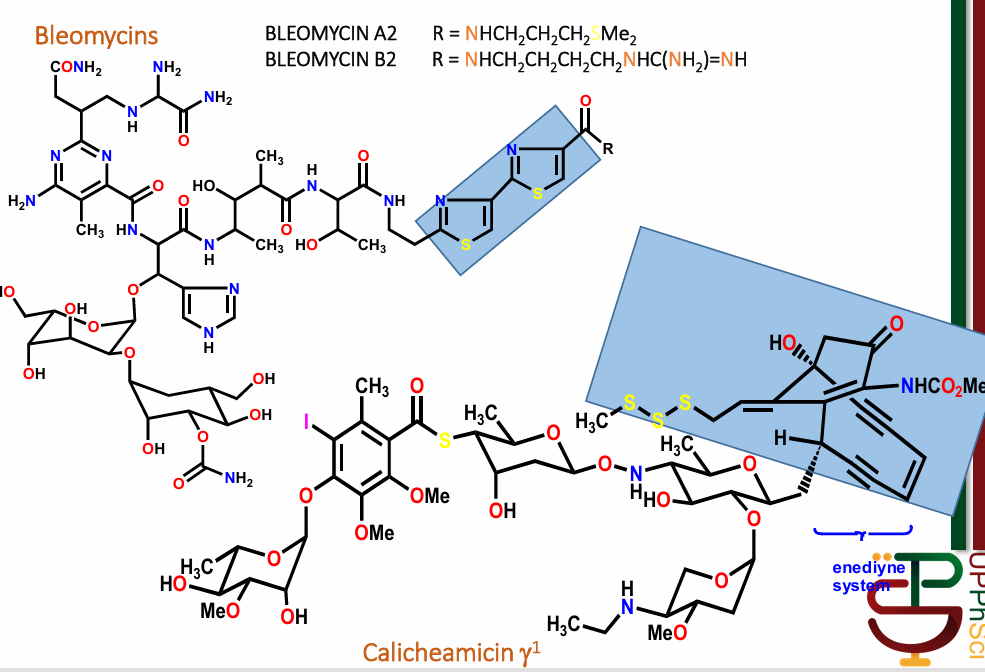
Chain Cutters
long, polypeptide antibiotics
ex: bleomycin, calicheamicin
highlighted portions generate free radicals within DNA structure
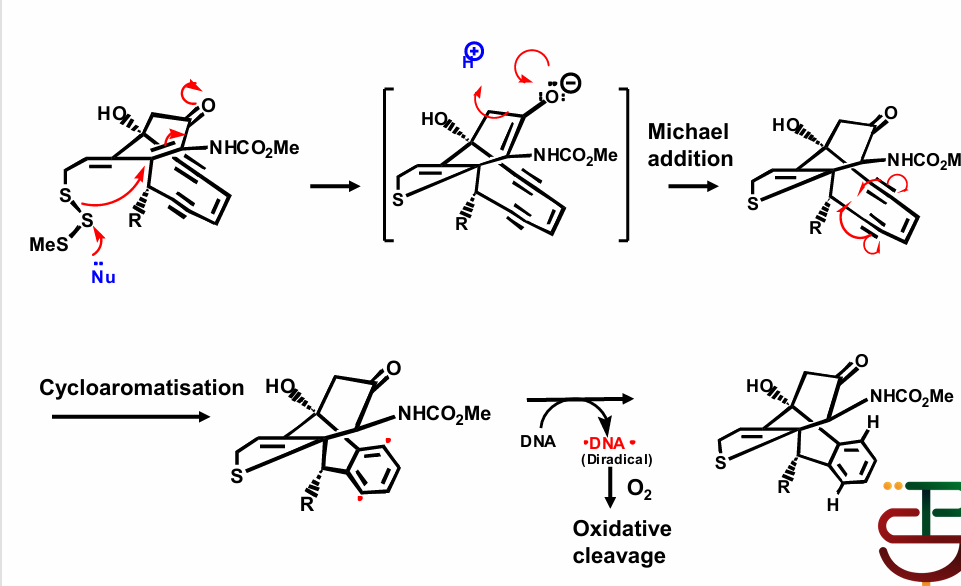
Mechanism:
Nucleophile attacks sulfur, causes electron rearrangement and breaks aromaticity
involves generation of diradical to regenerate aromaticity
Causes DNA to form diradical, resulting in oxidative cleavage
Alkylating Agents
old school, but still work (though are quite toxic)
Cell Cycle Non-Specific
tend to ironically cause secondary cancer due to damage
contain highly electrophilic groups
form covalent bonds to nucleophilic groups in DNA
particular target: 7-N of guanine (same as previous alkylating)
prevent replication and transcription via irreversible structure change
very effective; very toxic (strong electrophiles tend to be toxic to body, ex: alkylating proteins)
Alkylating Agents Mechanism
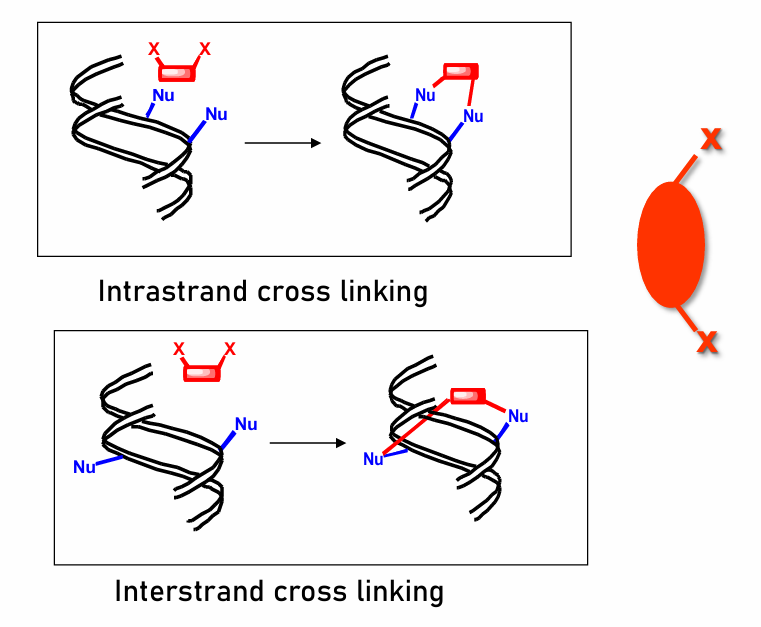
cross-linking
have good leaving group attached (generally two)
electrophile is generated when GLG leaves
Nucleophile in DNA attacks electrophile, forms covalent bond
generally prodrugs; leaving groups only leave when in body
Intrastand:
binds two ends to the same strand
generally for smaller alkylating agents
Interstrand:
binds two ends of two different coiled strands
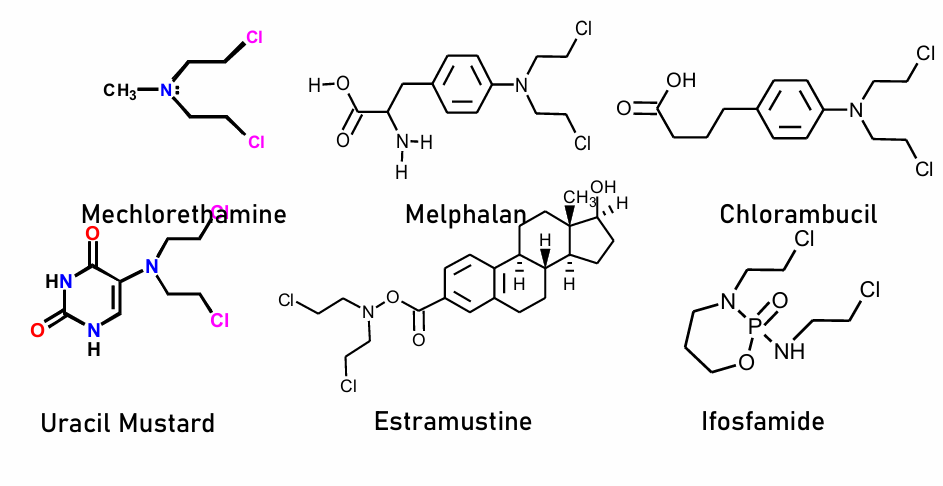
Alkylating Agents Examples
Nitrogen Mustards:
mechlorethamine
melphalan
chlorambucil
Uracil Mustard (most effective because cell recognizes it as uracil and it can go through the uracil transporters; then triphosphorylated)
estramustine
ifosfamide
Phosphamide Activation
type of nitrogen mustard
starts as cyclophosphamide
relatively nontoxic
hydroxylated by Cytochrome P450 enzymes into 4-hydroxyclyclophosphamide
then transforms in aldophosphamide
then converted to phosphoramide mustard (acrolein group leaves)
Nitrosoureas
also alkylating agents
highly lipid soluble, can cross BBB
used for brain tumor, meningeal leukemia
ex: Lomustine, Carmustine, Streptozocin (also used to induce diabetes in rats)
Mechanism:
initially transformed, electrophile activated by CYP450 enzymes
Electrophiles are attacked by C-G DNA Bases
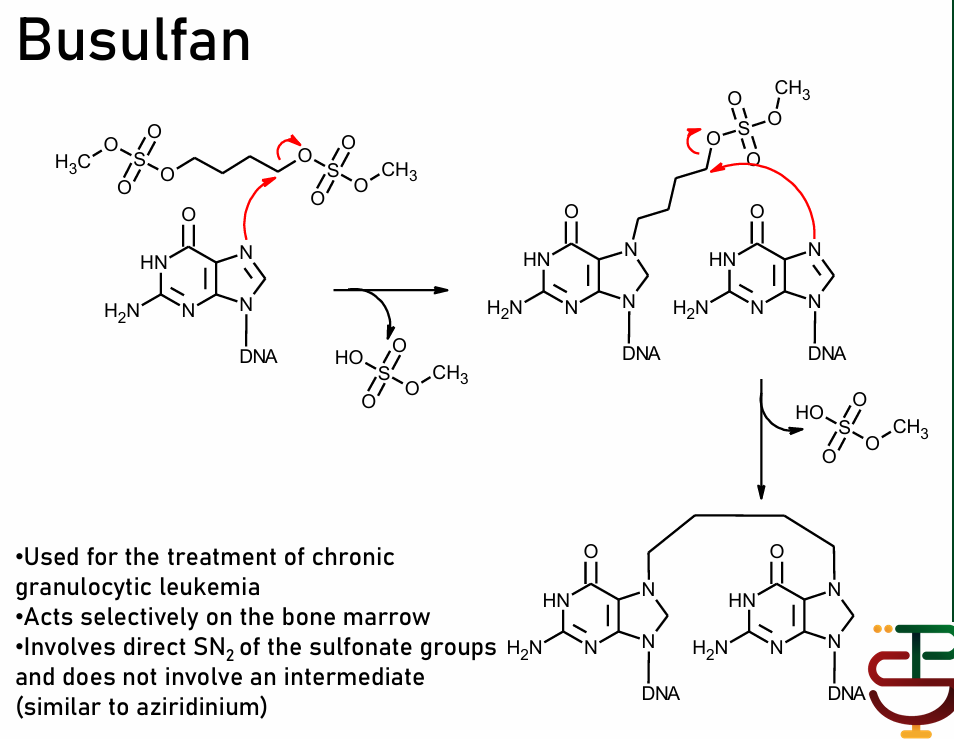
Busulfan
not processed by enzymes as prodrug; self-destructs to form electrophile
via direct SN2 reaction with sulfonate groups
selective bone marrow targeter
Platins
one of the most prescribed but most expensive cancer treatments
contains platinum in the structure
effective intrastrand cross-linker (all previous were interstrand)
ex: Cisplatin, Carboplatin, Oxiplatin
favors guanine residue
used for testicular and ovarian cancer
Mechanism:
LGs are attached to Pt
Water attacks Pt, kicks off LG
DNA Nu attacks Pt, kicks off water
process repeats for second DNA Nu
ends with intrastrand crosslink
Dacarbazine and Procarbazine
processed by CYP450 to form electrophile
complex structures, but end as CH3+ electrophile only (methyl cation)
not cross-linkers, only alkylating agent (methylates with cation)
for melanoma and Hodgkin’s Lymphoma
Mitomycin C
undergoes biotransformation by reduction
forms electrophile and again, is attacked by DNA
forms alkylation and interstrand crosslinking
prefers guanine residues
Antimetabolites
competitive inhibitors; prevent synthesis of DNA by reducing metabolite count
Ribonucleotide Reductase Inhibitors
Adenosine Deaminase Inhibitors
Thymidylate Synthase Inhibitors
Dihydrofluorate Reductase Inhibitors
DNA Polymerase Inhibitors
Purine Antagonists
Ribonucleotide Reductase Inhibitors
Ribonucleotide Reductase converts Ribonucleotide to Deoxyribonucleotide
Ribonucleotide: monomer for RNA
Deoxyribonucleotide: monomer for DNA
inhibition = no starting material for DNA synthesis
ex: hydroxyurea
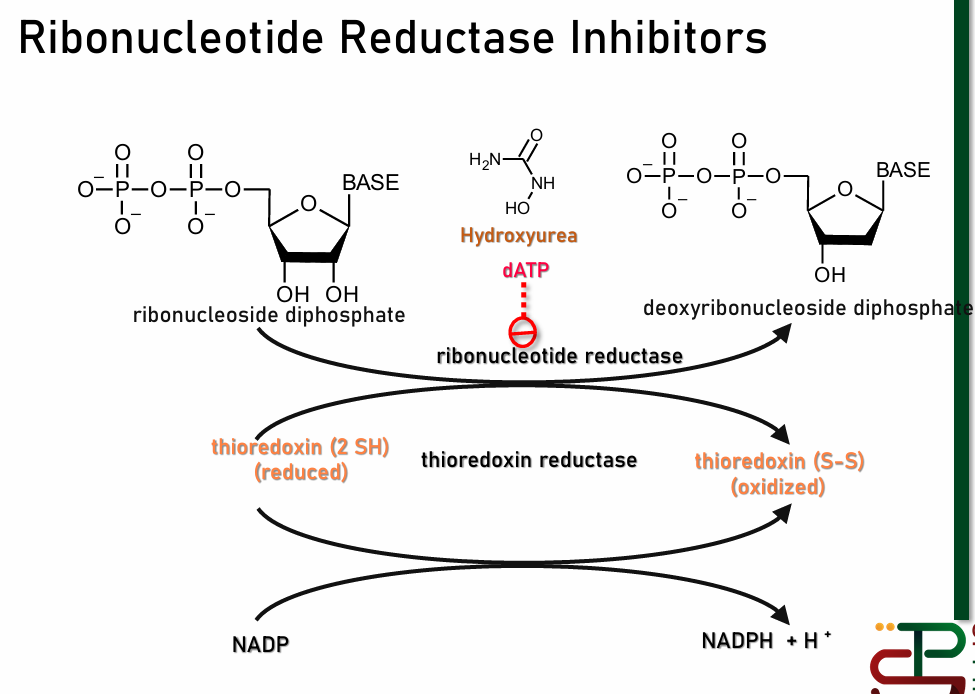
Note: due to this mechanism, thioredoxin reductase inhibitors will give similar effect
Adenosine Deaminase Inhibitor
AMP Deaminase converts AMP to IMP (Inosine Monophosphate)
IMP is an intermediate for AMP, GMP, etc which are used for DNA Synthesis
Also may become degredation product
prevents DNA synthesis by reducing intermediates
ex: Pentostatin
Note: no approved drug yet
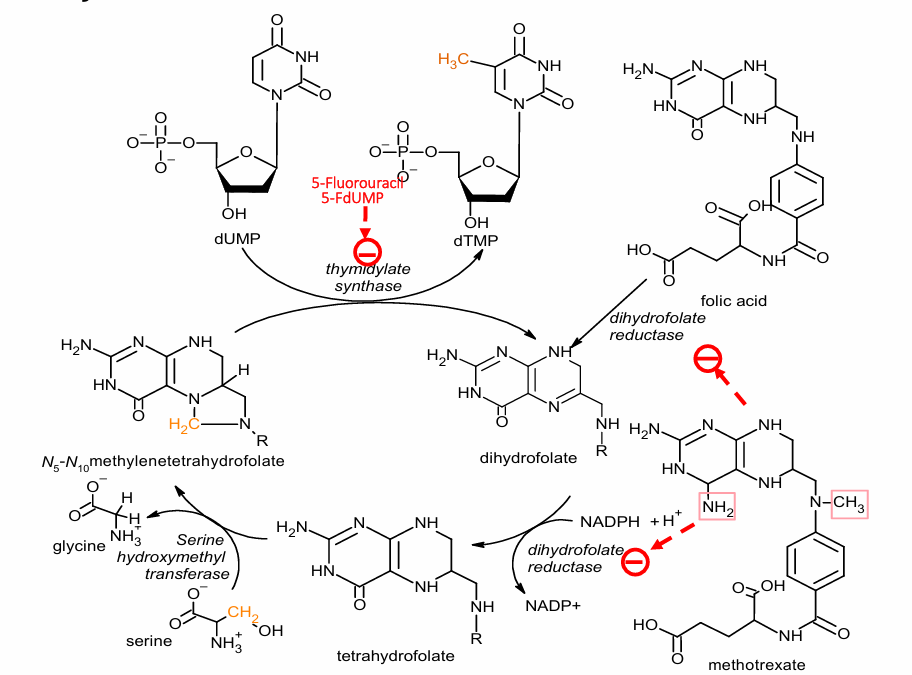
Thymidylate Synthase Inhibitor
converts deoxyuridine monophosphate (dUMP) to 5-deoxythymidine monophosphate (5-dTMP)
important for process of converting uracil (for RNA) into thymidine (for DNA)
ex: Raltitrexed, 5-fluorouracil
5-fluorouracil is converted to 5-fdUMP form, then competitively binds to enzyme
Dihydrofolate Reductaste Inhibitors
conversion of uracil to thymidine involves methylation
methyl group comes from tetrahydrofolate (Vitamin B9)
turns into dihydrofolate, must be reduced back into tetrahydrofolate
inhibition = no methylation of uracil = no thymidine
ex: Methotrexate (appears similar to folic acid); non-selective
because N is already
DNA Polymerase Inhibitors
Nucleoside Antimetabolites:
purine or pyridine looking sugar bases (nucleosides)
exist as prodrugs: body turns them into triphosphates
bind to DNA Polymerase instead of actual nucleotide base; prevents continuation as new bases can not attach to drug
also not de-oxy (has extra OH group); prevents continuation
ex: cytarabine, gemcitabine, fludarabine
Purine Antagonists:
appear like purine bases (no sugar)
processed by body from prodrug into active form
Note: both have to be activated into triphosphate form
Hormone Based Therapy
often steroidal agents (except GnRH Agonists)
Glucocorticoids
Estrogens
Progestins
Androgens
Gonadotropin-Releasing Hormone (GnRH) Agonists
Antiestrogens
Antiandrogens
Aromatase Inhibitors
Adrenocortical Suppressors
Note: receptor locations are either on cell surface (cascade to nucleus), or on nucleus
Glucocorticoids
mimic cortisol
taken orally for leukemias and lymphomas
ex: Prednisolone, Prednisone
Estrogens
different from androgens because they are aromatic
act as antagonists to counter androgen/testosterone, for testosterone-based/prostate cancers (ex: testicular cancer)
also act as antimetabolites for estrogen
lipophilic, intracellular; steroidal receptors are in nucleus, bind to DNA
receptors act as Transcription Factors
upon activation, dimerizes, adds to co-activator protein, then acts as T-Factor
ex: ethinylestradiol, diethylstilberstrol, diethylstilbestrol biphosphate, estramustine phosphate
Product is either another receptor or enzymes needed for hormone synthesis
Antiestrogens
estrogen antagonists
generally: antagonists are larger than competition
due to large locking side chain
can displace estrogen due to having higher affinity for enzyme
locks enzyme into inactive state
transcription does not proceed
for breast cancer, endometrial cancer, etc.
ex: Fulvestrant
Progestins
emulate progesterone
non-aromatic, so no longer estrogen
for breast cancer, endometrial cancer, etc.
ex: medroxyprogesterone acetate, megestrol acetate
Androgens
non-aromatic, differentiates from estrogen
Androgens can help treat breast cancer, ovarian cancer, etc. (estrogen dependent cancers)
ex: fluoxymestrone, testosterone proprionate, testolactone
Antiandrogens
antagonists for androgens that are not estrogens
for testicular, prostate, androgen dependent cancers
ex: biclutamide (very selective for androgen)
Gonadotropin Releasing Hormones (GnRH) Agonists
Gonadotropin Releasing Hormones triggers release of luteinizing hormone from pituitary gland
goes to gonads; triggers release of sex hormones
agonists mimic GnRH (are also peptides)
used to produce the sex hormone to counter the cancer (ex: estrogen for prostate cancer)
long-term effect: prolonged use = desensitization
leads to drop in luteinizing hormone
leads to eventual drop in sex hormone release
used for advanced prostate cancer
ex: Leuprolide, Goserelin
Aromatase Inhibitors
2nd line treatment for estrogen-dependent breast cancer (resistant to tamoxifen)
antimetabolite for hormone
Androgen is nonaromatic, Estrogen is aromatic
aromatase makes androgen aromatic; part of estrogen conversion
is part of CYP450
androgen → 4-hydroxyandrostenedione → estrogen
aromatase turns 4-hydroxy into estrogen
inhibition decreases estrogen synthesis
have reversible and irreversible inhibitors
for estrogen dependent cancers
the reversible ones are competitive
Reversible:
competitive
ex: aminoglutethimide, letrozole
Irreversible:
ex: 4-hydroxyandrostenedione, exemestane
Adrenocortical Suppressors
for adrenocortical tumors
limit the action of adrenocorticotropic hormone (ACTH)
helps treat cancers that form on adrenal gland
ex: mitotane
Target: Structural Proteins
tubulin
tubulin is the structural proteins in microtubules
important for mitotic spindle
inhibition of tubulin polymerization OR depolymerization will eventually lead to cell death
vinca alkaloids inhibit polymerization (ex: vinblastine, vincristine)
paclitaxel inhibits depolymerization
generally function only during mitotic phase
will generally spell apoptosis if can not depolymerize
cell needs to dissolve spindles after separation
Target: Signaling Pathway
for growth factor signaling cascades
tyrosine kinases dimerize to activate signaling pathway upon GF binding
Activates RAS protein which starts cascade that ends with gene transcription
Monoclonal Antibodies:
bind to either GF or GFR
Protein Kinase Inhibitors:
competitive inhibitors for phosphorylating substrate
must enter cell to work
Farnesyl Transferase Inhibitors:
essentially inhibit RAS protein by blocking RAS binding
Monoclonal Antibodies
Very specific, will not work otherwise
ex: Trastuzamab
HER2 receptor
for certain breast cancers (HER2 positive)
induces immune response upon binding
induces downregulation of receptor
Protein Kinase Inhibitors
Major Classes:
Tyrosine Kinases
Serine/Threonine Kinases
Histidine Kinases
Main Type of Inhibitors:
Type I: Active Conformation Binders
Bind to active dimerized receptor
prevent actual substrate from getting in
ex: Gefitinib - for chronic myeloid leukemia; first to target unique molecular structure for cancer (competes for ATP; extra region binds to hydrophobic region as anchor)
Type II: Inactive Conformation Binders
Bind to inactivated receptor
stabilize inactive form
Ex: Imatinib - first marketed PKI, used for chronic myeloid leukemia
Farnesyl Transferase Inhibitors
FT instrumental to GF cascade
RAS Protein only attaches to membrane when farnesylated
needs to be membrane-bound because recepto
inhibits RAS attachment
stops signaling cascade of Growth Factor
Miscellaneous Enzyme Inhibitors:
Matrix Metalloproteinase Inhibitors
Cyclooxygenase Inhibitors
Proteasome Inhibitors
Histone Deacetylase Inhibitors
Matrix Metalloproteinase Inhibitors
MMPs dissolve ECF
inhibition prevents dissolution, inhibits angiogenesis
cancer can not access nutrients
ex: Marimastat, Prinomastat
resemble peptides to attach to proteinases
are transition state inhibitors (higher affinity)
Cyclooxygenase-2 Inhibitors
COX-2
Inflammation, pain
Celecoxib
a symptomatic treatment, doesn’t treat cancer, but does treat resulting bad symptoms
Proteosome Inhibitors
more novel
inhibition can promote apoptosis
proteosome is important in many cell cycle steps
Proteosome: protein trash can
often unfolds or breaks misfolded proteins (tagged by ubiquitin)
P53 Protein: tumor suppressor protein
if discard/thrown to proteosome too much, tumors happen
Inhibition of proteosome decreases uncontrolled P53 degradation
Halts cell proliferation
targets myeloma
Proteosome Inhibition effects
prevents angiogenesis; metastasis
inhibits breakdown of some antibodies
can cause apoptosis
can inhibit necrosis factor kB
eventually causes folded protein response? = apoptosis
Histone Deacetylase Inhibitors
epigenetic target
HDs remove acetyl group from histones, make them positive
Negative phosphate of DNA then winds around it
if wound, is not expressed (silences)
If too much Tumor Suppressor Genes are suppressed = cancer
Action: inhibits deacetylase, freeing Tumor Suppressor Genes
resemble epsilon-amino area of Lysine
Vorinostat: first FDA approved in this class (hits HDAC1, HDAC2, HDAC3, HDAC6)
can also promote immune response
Synthetic Agents
Thalidomide:
notoriously teratogenic (baby with no limbs)
immunomodulator for multiple myeloma
repurposed: from nausea and morning sickness for pregnant woman to drug for cancer
Revamid
d
Actimid:
d
Arsenic Trioxide:
orphan drug (no category it can be put under)
effective for certain leukemias
suspected to target mitochondria via generation of ROS
Natural Products
promising use
Cephalostatin & Pancra:
potent for
not yet in clinical trials
Protein Therapy
Angiostatin
Endostatin
Alpha-Interferons
Complement immune response
Upon injection; promotes immune ability against cancer cells
TRAIL (Tumor Necrosis Factor Related Apoptosis Induced Ligand)
Short polypeptides
Not really in investigation anymore
L-asparaginase
Some cancers rely on amount of asparagine
Asparaginase deprives cancers of them
Cell Targeted Therapy
Antibody-Drug Conjugates
ADEPT
GDEPT
AFN
selective for certain target cells/cancer cells
hoping certain cancers have unique signals
if it happens that cancers only overexpress some proteins (but don’t make unique ones), then this may not work as well
Antibody-Drug Conjugates
drug is attached to an antibody
antibody is selective for cancer antigen
Antibody connects to cancer cell, bringing drug with it
Drug now in close proximity with cancer cell; can enter cancer cell
Antibody Directed Enzyme Prodrug Therapy (ADEPT)
To avoid premature activation: conjugates enzyme with antibody
Enzyme connects to antibody connects to target
Injects drug into blood
Drug will be activated by enzyme when at target site
Still has some risk of going somewhere else and being toxic
Gene Directed Enzyme Prodrug Therapy (GDEPT)
gene encoding for drug activating enzyme is administered to cancer cells
cancer cells begin producing enzyme
prodrug is administered, activated upon entering cell
nontoxic to all other cells, only become toxic upon entering cell
Aptamer-Functionalized Nanoparticle
no approved yet
Aptamer: single stranded DNA/RNA that is highly selective for
Plasmid DNA: codes for proteins that induce apoptosis
may also contain a cytotoxic agent
Aptamers bind to cell and it is taken in via receptor mediated endocytosis
Activates and induces apoptosis
Photodynamic Therapy
given light sensitive drug
drug is distributed to oxygen-hungry cancer cells
laser shone on cancer spots, activates drug; kills
less effective in oxygen deficient tumor environment
less effective for large tumors
Photothermal Therapy
administers chemotherapeutic agent
it distributes
shoots it with light and heat laser
Combination Therapy
rational drug combinations
cancer is very resilient, adaptive, and has multiple mechanisms
as such, combination therapy with several rationally combined agents can be more effective
challenge is to pick correct combination
Ex:
targeting signaling pathway, and then targeting its reactivation/alternative step
Maximal driver pathway inhibition
Enhanced synthetic lethality
Collateral lethality: gene therapy?
Alternating treatment for addiction or resistance
Targeting heterogeneity and drug resistant pools (combo of drug for sensitive and drug for resistant; ex: selective + nonselective combo)
Targets Immune Cell Function
Modulating tumor and environment
ICB (Antibody) + Chemo + Radio
Sensitizing tumor cells to immunotherapy
Modulating microbiome
Neoadjuvant therapies
If just chemotherapy, may ignore what happens outside the cell that may contribute to cancer
Experimental Combination Therapy
chemo + Antibody
cytokine + antibody
Inhibitors + Nucleic Acids
T-Cells + chemo
Aptamers very usefu