(pt 3) exam #5 - heme II (cls 546)
1/117
Earn XP
Description and Tags
fibrinolysis + thrombophilia (updated 5/9 for final)
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
118 Terms
how do we control coagulation?
inhibitors, cofactors, feedback loops, blood flow (vasoconstriction)
where do all the activated cofactors go?
cleared by the liver
coagulation regulatory mechanisms
Primary regulators
AT ; TFPI ; Protein C
Also regulating coagulation:
Thrombomodulin (TM )
Endothelial protein C receptor (EPCR)
Heparin cofactor II
Protein S
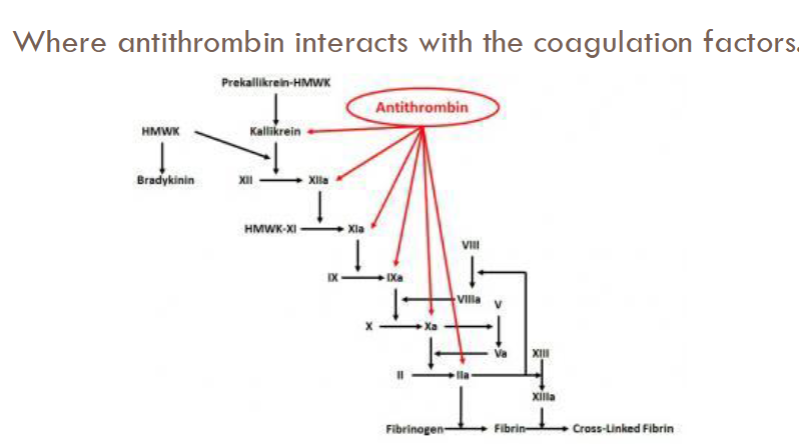
antithrombin (AT)
Plasma proteinase inhibitor (serpin)
Activation of AT by vessel wall heparan sulfate proteoglycans (HSPGs)
Activity enhanced 3 to 4 fold by heparan sulfate (glycosaminoglycan found in ECs)
Most important inhibitor of serine proteases
Inactivates thrombin and other enzymes responsible for thrombin generation
indirect thrombin inhibitors (list)
Heparin (UFH, HMWH)
Low molecular weight heparins (LMWH)
Enoxaparin
Dalteparin
Tinzaparin
Fondaparinux
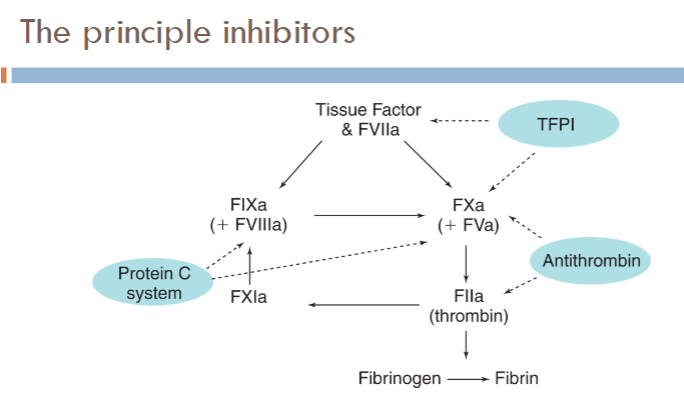
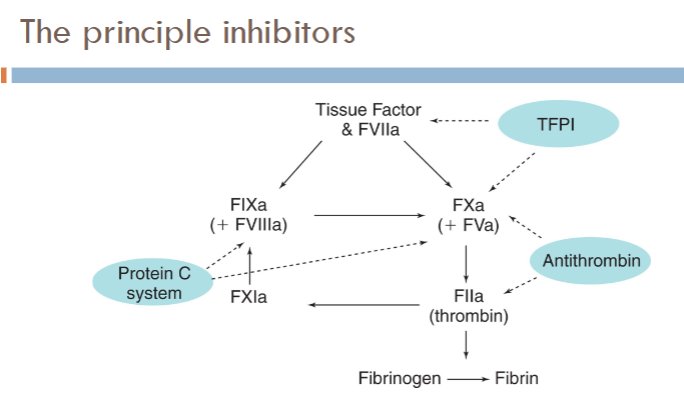
what are the principle regulators/inhibitors of coagulation?
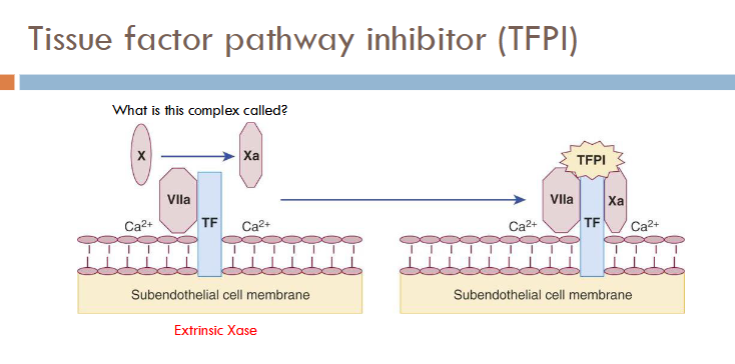
Tissue factor pathway inhibitor (TFPI)
Anti-thrombin (AT)
Protein C pathway (knocks out intrinsic pathway)
tissue factor pathway inhibitor (TFPI) pathway picture
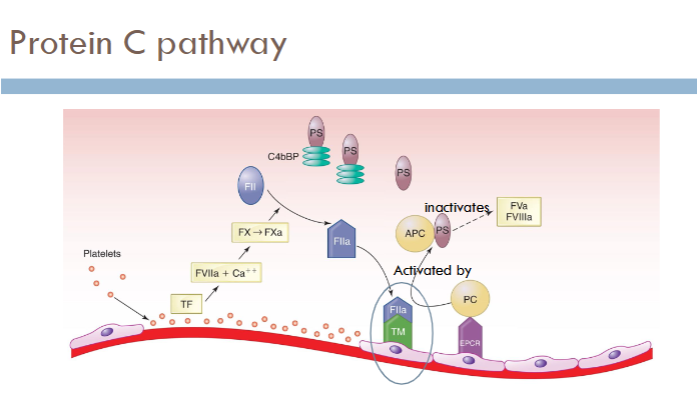
(fibrinolysis) protein C pathway
how is fibrin formed?
thrombin cleaves off FPA and FPB from fibrinogen
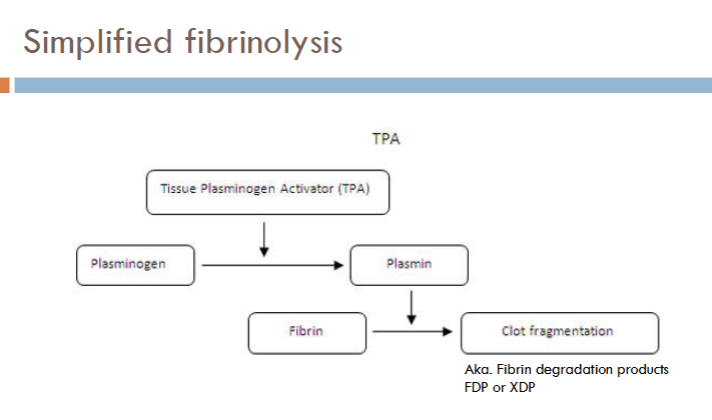
what is fibrinolysis?
process of digesting and removing fibrin
how is fibrinolysis activated?
activated in response to cascade activation and fibrin formation
what happens if you have excessive fibrinolysis? inadequate fibrinoylsis?
excessive = bleeding
inadequate = excessive clotting
(players of fibrinolysis) components
Plasminogen (PLG)
Plasmin (PLN)
(players of fibrinolysis) activators
Tissue plasminogen activator (tPA)
Urokinase type plasminogen activator (uPA)
Factor XII
Prekallikrein (PK)
(players of fibrinolysis) inhibitors
Plasminogen activator inhibitor 1 (PAI-1)
A2--antiplasmin (AP)
Thrombin activated fibrinolysis inhibitor (TAFI)
C-1 esterase inhibitor (C-INH)
α2 – macroglobulin (α2-M)
(players of fibrinolysis) cell surface receptors
uPA Receptor (uPAR)
Annexin 2
Low density lipoprotein receptor like protein (LRP)
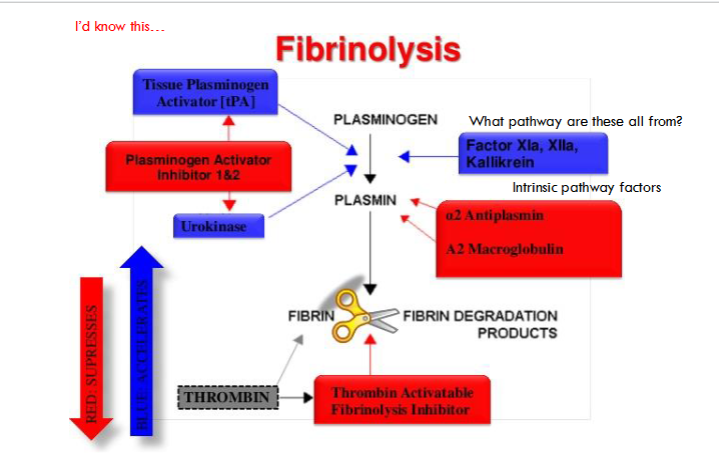
fibrinolysis diagram
(fibrinolysis) plasminogen (PLG)
Circulates as single polypeptide chain
Synthesized by the liver
Activated by plasminogen activators (PAs) to plasmin (PLN) by proteolytic cleavage
Substrates include:
Fibrin, fibrinogen, V, VIII, VWF, complement, several hormones, and platelet surface receptors
(fibrinolysis) tissue-type plasminogen activator (tPA)
Released from ECs in vivo as single chain molecule (sctPA)
Does NOT circulate as a zymogen
Fully active in single chain form
Binds to fibrin surface
Intracellular stores released when stimulated
Thrombin, BK, histamine, exercise, DDVAP, and so on
Can also bind to EC surface via receptor--annexin
Maintains fibrinolytic potential on undamaged vascular surfaces
(fibrinolysis) urinary-type TPA (uPA) / urokinase
Found in urine and plasma
Functions mainly in tissue
Digests extracellular matrix
Enables cell migration
Important in wound healing, inflammation, cancer metastasis
Converted to two chain active form by PLN, XIIa or kallikrein
Does not need fibrin as cofactor
what are the 2 major plasminogen activators? how else is plasmingoen activated?
tPA, uPA (urinary type plasminogen activator/urokinase)
also activated by contact factors (XIIa, Xia, kallikrein)
(fibrinolysis) exogenous activators
Not used in US
Streptokinase
Derived from beta-hemolytic strep
Staphylokinase (SAK)
Produced by S aureus
Alteplase--tPA--recombinant tPA
(fibrinolysis) plasmin
Responsible for degradation of fibrin (or fibrinogen)
Distinct protein fragments produced
Fibrin degradation products (FDP)
Sites of plasmin cleavage
Similar in fibrin and fibrinogen
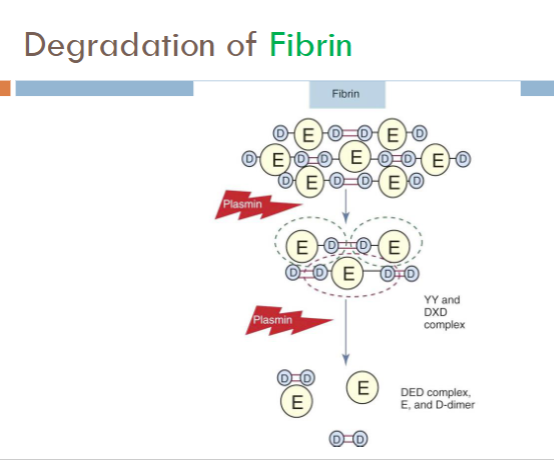
fibrinogen degradation
Products formed from fibrinogen cleavage
Fragment X (trinodular structure)
Fragment Y (binodular D + E)
Fragments D and E (uninodular)
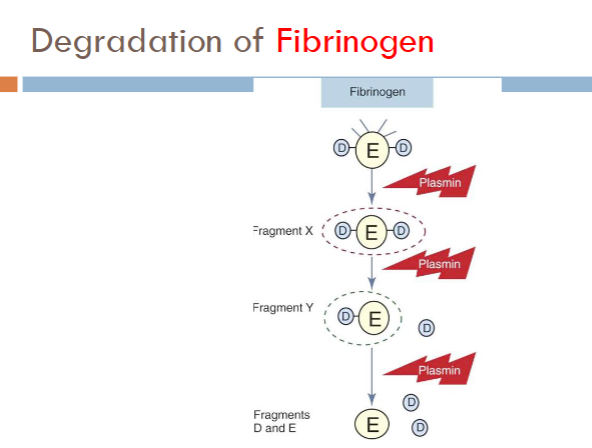
fibrin degradation
Products formed from fibrin cleavage
Essentially the same as fibrinogen, except:
XIIIa-crosslinked fibrin releases
D-Dimer
Larger molecules of varying composistion
DD/E; YD/DY; YY/DXD
systemic effects of plasmin
If not controlled, plasmin can degrade
Fibrinogen V, VIII, and XII
Components of kinin and complement systems
fibrinolytic inhibitors
Prevent systemic proteolysis
Act at the:
PLG activation step
Plasminogen activator inhibitors/PAI
Directly on plasmin
a2-antiplasmin
a2-macroglobulin
Directly on thrombin
Thrombin-activatable fibrinolysis inhibitor (TAFI)
(fibrinolytic inhibitors) plasminogen activator inhibitor (PAI-1)
Primary physiological inhibitor of tPA and uPA
Acute-phase reactant
Increases during inflammation, stress
Decreased levels shift hemostatic balance toward hypercoagulability
(fibrinolytic inhibitors) thrombin-activatable fibrinolysis inhibitor (TAFI)
Activated by the thrombin/thrombomodulin complex
Inhibits fibrinolysis
Downregulates the cofactor functions of fibrin by cleaving lysine residues
Interferes temporarily with the interaction of tPA and PLG with fibrin
TAFI has short half-life
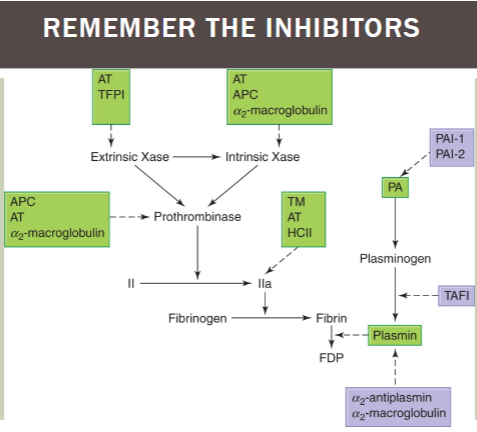
fibrinolysis inhibitors / activators chart
extrinsic Xase: inhibited by AT, TFPI
intrinsic Xase AND prothrombinase: inhibited by AT, APC, a2-macroglobulin
factor IIa (thrombin): inhibited by TM, AT, HCII
plasminogen → plasmin: inhibited by PAI-1/2; TAFI; a2-antiplasmin/a2-macroglobulin
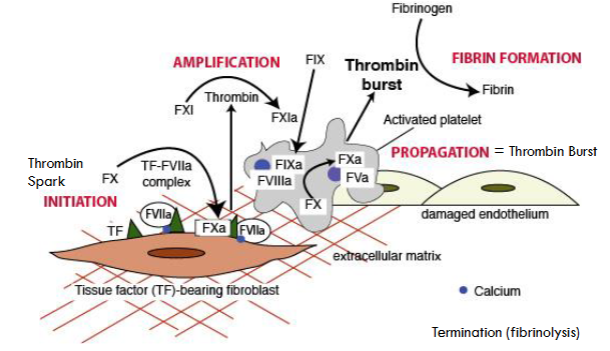
what are the phases of cell-based coagulation theory?
initiation (thrombin spark)
amplification
propagation (thrombin burst)
fibrin formation
termination (fibrinolysis)
difference in coag cascde and cell based theory
coag cascade
extrinsic/intrinsic pathways work independently & meet at common end point
both make large amt of thrombin to form clot
coag factors control rate of coagulation, cells = phospholipid surface
cell based
extrinsic pathway initiates process—generates small amt of thrombin @ thrombin spark
followed by intrinsic pathway—works on platelets to produce thrombin burst (no role of factor XII)
cells control the coagulation

difference between thrombus and clot?
Clot = forms on the outside of a vessel on tissue (extravascularly)
Thrombus = forms on the inside of the lumen of a vessel (intravascularly)
what mechanisms are involved in abnormal clot formation? (3)
lack of inhibitors to clotting
stimulation of clotting
problems w activators or inhibitors to fibrinolysis
what is ischemia?
lack of blood flow when a clot blocks a blood vessel
what is necrosis?
cell/tissue death due to blocked vessels
what is an embolism?
clot or thrombus moves from site of creation to another site where it blocks blood flow
what is a thromboembolism (TE)?
thrombus that moves from site of creation to another site where it blocks blood flow
white thrombi
ARTERIAL THROMBI
Composed primarily of platelets and fibrin
Few leukocytes and erythrocytes
Usually form at:
Regions of disturbed blood flow
Sites of damage to endothelium
Atherosclerotic plaques
Plaque
Composed of lipids, fibrous connective tissue, macrophages, and smooth muscle cells
how are white thrombi (aterial thrombi) formed?
created when plaque ruptures
Exposing thrombogenic material in subendothelium to blood
Activation of platelets and plasma coagulation factors
Fibrin formation → thrombus
Can lead to embolization
Myocardial or cerebral infarction
Therapy
Platelet-inhibiting drugs (aspiring, clopidogrel, ticlopidine) or thrombolytic therapy
risk factors for developing ARTERIAL thrombi
Traditional risk factors:
Hypercholesterolemia ; hypertension
Smoking ; physical inactivity
Obesity ; diabetes
More recently recognized risk factors
Hyperhomocysteinemia, ↑Lp(a), oxLDL
how to detect arterial thrombi?
Standard hemostasis--not sensitive or specific
"new" potential tests:
Sensitive, still not specific
Hyperhomocysteinemia
↑ Elevated Lp(a)
↑ fibrinogen
↑ D-dimer
↑ PAI-1 or ↓ t-PA
↑ high sensitivity CRP (hsCRP)
what is the role of inflammation in clots/thrombi formation?
increases vascular occlusion (often precedes it) increase inflammation increases vascular risk
if arterial thrombi are called white, what are venous thrombi called?
red thrombi
what are venous thrombi composed of?
RBCs
red/venous thrombi
Composed primarily of RBCs
Most commonly occur in veins of lower limbs
Thrombophlebitis
Superficial veins of legs
Usually benign
Deep vein thrombosis (DVT)
Deep veins of legs
Distal thrombi--less serious than thrombi is proximal veins (popliteal, femoral, oriliac)
Venous thrombi lead to serious potential complications
Pulmonary embolism (PE)
DVT → PE: VTE
how do venous (red) thrombi form?
Occurs when activation of blood coagulation exceeds the ability of the natural protective mechanisms to prevent fibrin formation
Laboratory diagnosis difficult
Objective diagnostic tests--visualization of thrombus
risk factors for VENOUS thrombi
Venous stasis
Vessel wall damage
Factor V Leiden (FVL) and APC resistance
Deficiency of protease inhibitors (AT, PC, PS, HCII)
Elevated prothrombin levels (Prothrombin 20210)
Antiphospholipid antibodies
Hyperhomocysteinemia
Decreased fibrinolytic activity
Malignancy ; surgery
Miscellaneous
Advanced age, obesity, blood type, pregnancy, OC/HRT, smoking, hypertension, hyperlipidemia
**the more risk factors, the most likely the clot forms
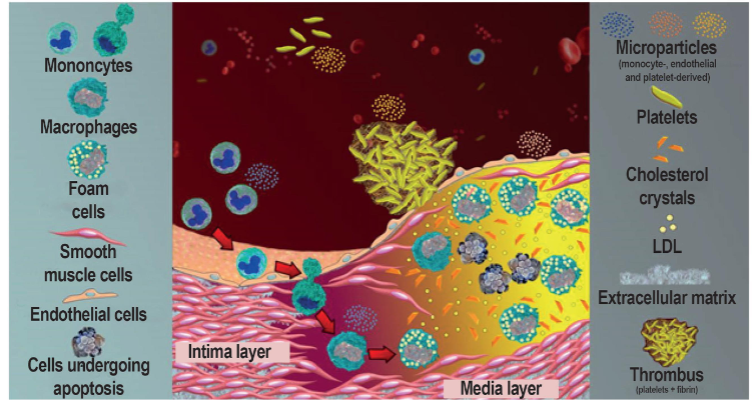
microparticles in thrombosis
Cell-derived products which play key role in thrombus formation
Released from cell membrane during apoptosis or activation
Several roles
Have procoagulant potential
Play role in inflammation, angiogenesis, and immune response
Promote anticoagulant functions and fibrinolytic response
Derived from platelets, ECs, RBCs, WBCs, cancer cells
Differ in expression of phosphatidyl serine and/or TF
Controversy--cause of consequence of thrombosis
what makes someone hypercoagulable?
when more procoagulant activity is happening than anticoagulant activity
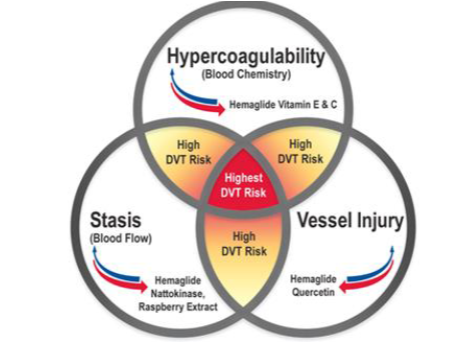
virchow’s triad
hypercoagulability, stasis, and vessel injury
made by rudolph virchow to develop/propose mechanism for PE
what are the 2 genes linked to hyperhomocysteinemia? how is it treated?
genes: cystathionine B synthase (CBS) + methylenetetrahydrofolate reductase (MTHFR)
treatment: vitamin supplements (folic acid/B6/B12), dietary changes
thrombophilia
any disorder with an increased risk fo VTE ; aka hypercoagulability
hereditary thrombophilia
Most inherited alterations associated with:
↑ in procoagulant potential
↓ in natural inhibitors of clotting
Abnormalities of fibrinolysis or platelet activation
Inherited thrombophilic defect
Often requires some other (acquired) risk factor—surgery, pregnancy, immobilization, estrogen therapy, obesity, trauma, infection
Combination of hereditary predisposition and acquired predisposition
Accelerates and exaggerates thrombotic process
"Multiple hit" theory of thrombosis
indicators of hereditary thrombophilia
Usually associated with venous thrombosis
Suggested by:
Venous thromboembolism at a young age (prior to 43)
Recurrent venous thromboembolism
Family history of venous thromboembolism
Thrombosis in an unusual site
Cervical or visceral vein
Retinal ; cerebral vein
most common inherited thrombophilias (5)
Antithrombin (AT or AT3)
PC
PS
Factor V Leiden
Prothrombin 20120 mutation
**account for 30% of initial DVT cases ; 50% normal levels assoc w increased risk
(thrombophilia) antithrombin deficiency pathophysiology
>200 different mutations described
Type I deficiency--quantitative deficiency
Type II deficiency—qualitative defect
Mutation of either heparin binding site or thrombin binding site
indications of hereditary AT3 deficiency
Incident or recurring VTE
Family Hx of VTE
Recurring miscarriage or pregnancy complications
Hx of arterial thrombosis (<50 yrs old)
Hx of arterial thrombosis in patients w/ no other risks for atherosclerosis
aquired antithrombin deficiency
Decreased production—severe liver disease
Increased consumption
Acute thrombosis, DIC
Increased clearance or loss
Nephrotic syndrome ; heparin
High dose OCs and late pregnancy
Increased FIB and coag factor activity (VII and X)
Decreased ATIII
laboratory diagnosis of antithrombin defiency
Functional assays
progressive AT assay
Quantifies the ability of AT to neutralize thrombin or Xa w/o Heparin
heparin cofactor assay
Measures the ability of anti-thrombin to bind heparin & neutralize thrombin or factor Xa
Antigenic assays (nonfunctional)
Immunologic--chromogenic
Differentiate type I and II (quantitative vs qualitative)
treatment of antithrombin deficiency
Heparin
AT concentrates
Prophylactic therapy usually unnecessary
If less than <30% AT3 present, may do oral anticoagulants (Coumadin. Eliquis, Pradaxa etc)
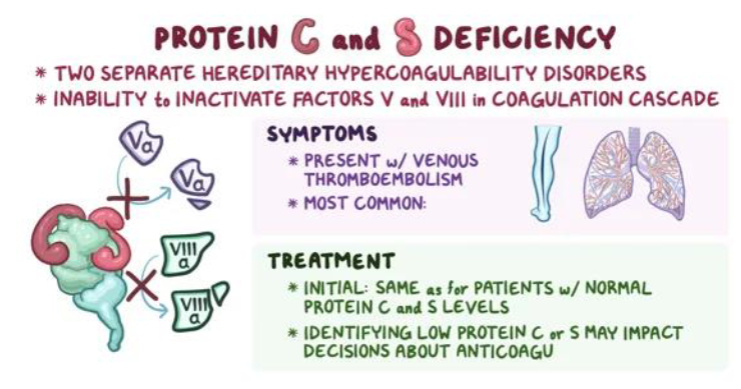
(thrombophilia) protein C deficiency pathophysiology
Reduction of PC to about 50% or normal predisposes to venous thrombosis
Autosomal dominant inheritance
6% of familial thrombophilia cases
>180 different mutations described
Type I deficiency--quantitative deficiency
Type II deficiency--qualitative deficiency
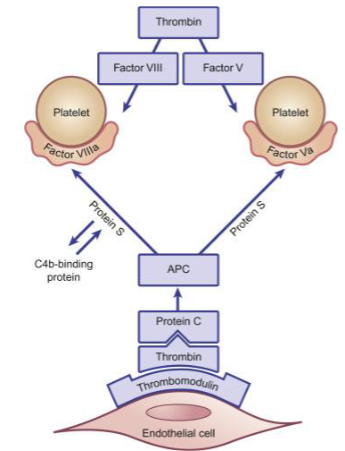
how is protein C activated?
binding of thrombin to thrombomodulin
what does activated protein C (APC) do?
degrades Va and VIIIa
what are the cofactors for protein C?
protein S and Ca2+
how does protein C deficiency work?
Predisposes to thrombosis because:
Decreased capacity to destroy FVa and FVIIIa
Results in an increased generation of thrombin and fibrin
Loss of cytoprotective effect (anti-inflammatory and anti-apoptotic) of APC binding to EPCR
Copperhead snake venom activates PC
(thrombophilia) protein S deficiency pathophysiology
Protein S circulates in 2 forms
Inactive form bound to C4b-BP – complement (60%)
Unbound or free form (40% )
Only free PS has PC cofactor activity
Absolute and relative amounts of free PS
Determined by plasma concentration of C4bBP
↓ free PS
Prothrombotic tendency due to inadequate APC inactivation of FVa and FVIIIa
DRW used in Protein S activity testing
(protein S deficiency) presentation, lab evaluation, treatment
Clinical presentation
Similar to that of PC deficiency
~6% of familial thrombophilia cases
Laboratory evaluation
Measure both total and free PS antigen levels
Immunologic assays
Functional assay based on ability to serve as cofactor for anticoagulant effect of APC
Treatment—similar to PC deficiency
(thrombophilia) activated protein C resistance pathophysiology
aka Factor V Leiden
In vitro—inability of activated protein C to prolong clotting tests when added to a test system
Due to a diminished ability to destroy FVa
In vivo—inadequate FVa inactivation
Increased production of thrombin and possibly thrombosis
cause of activated protein C resistance / factor V leiden
90% of cases of APCR due to FV Leiden
Arg→Gln mutation at AA #506
Mutant FVa protein resistant to APC inactivation because APC cleavage site is altered
10% of patients have alternate mutation in factor V
how does activated protein C work in factor V leiden?
if factor V is mutated, it becomes resistant to the inactivation by APC
leads to inability to stop coagulation = clot formation
(activated protein C resistance / factor V leiden) laboratory dx & treatment
Laboratory Dx
Requires both clot based functional assay (APCR) and PCR-based molecular assay for FVL
Treatment
Same as acute thrombosis
Prophylactic treatment--only if at increased risk or have recurrent thrombosis
(thrombophilia) prothrombin mutation 20210 pathophysiology
Glycine → alanine substitution in 3' untranslated region of the prothrombin gene
CAUSES INCREASED EXPRESSION OF PROTHROMBIN
Associated with a mild elevation of plasma prothrombin levels (115%-130%)
Increased risk of thrombosis due to:
Increased prothrombin causing increased thrombin generation
Accounts for ~18% of familial thrombophilia cases
testing for prothrombin mutation 20120
Molecular testing for the single point mutation is required
Other coagulation screen are unreliable
other players in thrombosis risk (5)
Heparin cofactor II deficiency
Inherited, DIC, liver disease
TFPI variant
TFPI gene mutation increased VTE risk
ABO blood groups
Type B and A1 have more VWF & VIII ; type O has less
VWF & VIII are acute phase reactants
Hyperhemocysteinemia (HC)
CBS or MTHFR gene mutation
Fibrinolytic disorders
Dysfibrinogen, decreased plasminogen or tPA, or PAI (excess)
list of the major anticoagulants (6)
unfractionated heparin
low molecular weight heparin
warfarin (coumadin)
dabigatran, bivalirudin (pradaxa, angiomax)
rivaroxaban, apixaban (xarelto, eliqus)
fondaprinux (arixtra)
mode of action for unfractionated heparin
inhibits XI, IX, thrombin
also inhibits Xa
mode of action for low molecular weight heparin
inhibits X & thrombin
mode of action for warfarin/coumadin
targets thrombin, VII, IX, X (2, 7, 9, 10)
vitamin K antagonist
mode of action for dabigatran, bivalirudin (pradaxa, angiomax)
direct thrombin inhibitor
mode of action for rivaroxaban, apixaban (xarelto, eliquis)
direct Xa inhibitor
mode of action for fondaprinux (arixtra)
indirect Xa inhibitor (binds to AT)
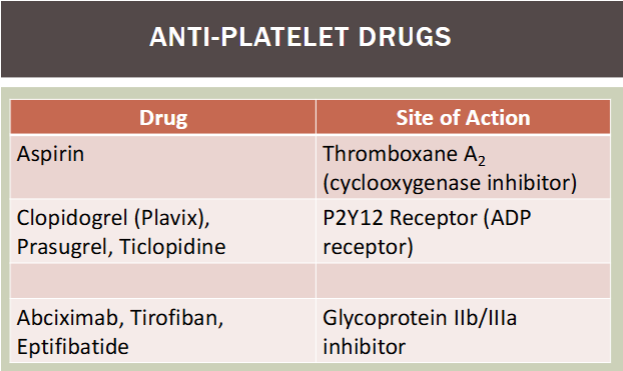
anti-platelet drugs / site of action
aspirin / thromboxane A2 (COX inhibitor)
clopidogrel (plavix), prasugrel, ticlopidine / P2Y12 receptor (ADP receptor)
abciximab, tirofiban, eptifibatide / glycoprotein IIb/IIIa inhibitor
heparin
Administered parenterally (IV or Sub-Q)
Does NOT have direct effect on blood clotting
Increases AT's ability to neutralize serine proteases
Extracted from porcine intestinal mucosa or bovine lung
UFH--MW between 5,000 and 30,000 Da
LMWH--MW between 4500-5000 Da
Prepared by enzymatic/chemical depolymerization of UFH
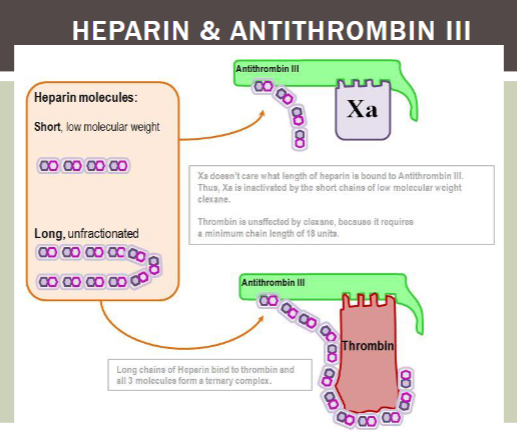
what test is used to monitor heparin therapy?
PTT/INR
standard/unfractionated heparin (UFH)
Catalyzes inhibition of most serine proteases
Influenced by "heparin-binding proteins" (acute-phase reactants) and high FVIII levels
LMWH & fondaparinux
Produces majority of its effect by catalyzing interaction of AT and FXa
More reliable pharmacokinetics because
Significantly less binding to heparin-binding proteins
Typically does not require routine laboratory monitoring
If monitoring needed (obese patients, renal disease, etc.)—use anti-FXa assay
what anticoagulant is monitored using the PT?
warfarin
(oral anticoagulants) coumadin drugs
Sodium warfarin or dicoumarol
Inhibit coagulation by interfering with vitamin K action in the liver
Blocks vitamin K-dependent carboxylation of target proteins
Resulting in release of nonfunctional molecules in the plasma
(γ carboxylated proteins)
Biologic effect—takes several days to be achieved
Half-life of preformed biologically active "normal" clotting factors
FVII disappears most rapidly (T½~6 hours)
what anticoagulants act through Anti-Xa activity?
Xarelto, Eliquis, Lixiana, (Rivaroxaban, Apixaban, Edoxaban)
what mechanism does Dabigatran or Pradaxa use to anticoagulate?
thrombin inhibitor
what does Clopidogrel or Plavix do?
platelet inhibitor through the ADP receptor (P2Y12)
how does aspirin inhibit platelet function?
arachidonic acid pathway
COX2 inhibitor thru TXA (thromboxane A2)
acquired thrombohemorrhagic conditions (list)
Acquired defects as common as inherited
Clinical presentation can range from thrombosis to bleeding--thrombohemorrhagic conditions
Acquired fibrinolytic defects
Anti-phospholipid antibodies (aPL)
Heparin-induced thrombocytopenia (HIT)
Flashback to HIT vs HAT
acquired fibrinolytic defects
30-40% of patients w thromboembolic disease
Have impaired fibrinolytic function
Seen post-surgery, coronary disease, OC, third trimester pregnancy, certain infections, drugs, malignancies
Increased plasma PAI-1 most common
+/- decreased t-PA
Long lasting inflammatory response
Elevated PAI-1 (acute phase reactant)
antiphospholipid antibody syndrome (APLS)
Include:
Lupus anticoagulant (LA)
Anticardiolipin antibodies (aCL)
Several subgroups--antibodies to PLs and PL binding proteins
Produced after certain infections, exposure to medications, in autoimmune disorders
Most common cause of acquired thrombophilia
5-30% of patients w VTE are positive for LA
antiphospholipid antibody syndrome (APLS) pathophysiology
Laboratory phenomenon
Prolongation of phospholipid-dependent clotting assays in vitro—hence term "anticoagulant"
Usually no bleeding diathesis
More often associated with increased risk of arterial and venous thrombosis
major clinical features of APLS
Venous thromboembolism (VTE)
DVT, PE
Arterial thromboses
Stroke, TIA, MI
Recurrent miscarriages
antibodies in APLS
Antigenic specificities of "aPL" antibodies
Usually IgG, can be IgM
Do NOT recognize PLs directly
Recognize phospholipid-binding proteins
Ex: B2 glycoprotein-I, prothrombin, V, VII, PC, PS, TFPI
aPL antibodies associated with:
Autoimmune disorders (SLE, Sjogren syndrome, RA)
Infections (malaria, parasitic)
Drugs (neuroleptics, quinidine etc)
aPL antibodies in the general population (~3-10%)
lab evaluation of APLS
immunoassays
Originally designed to detect ABY that recognize PL components
ELISA for anticardiolipin, ABS, anti-PS
New assays designed to detect ABS recognizing phospholipid cofactors (ex: anti-Beta2GPI)
coagulation assays
AB reacts w phospholipids in the test system and prolongs clotting
Sensitivity of assay influenced by reagent used
Could be an incidental finding w/ prolonged coag screen