Chemotherapy module 2
1/74
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
75 Terms
What are the 2 stages of the cell cycle?
Interphase: stage of growth prior to division (G1, S, G0, G2)
Mitosis: stage of cell division (Prophase, metaphase, anaphase, telophase, cytokinesis)
Describe the G0 phase
rest period, no cell division
Describe the G1 phase
Cells begin growing larger
Describe the S phase
Cells synthesis DNA
Describe the G2 phase
Cells grow even more and organise its contents in preparation for mitosis
Describe the M phase
cell division occurs (PMAT)
Describe prophase
DNA condenses forming chromosomes
Describe metaphase
nuclear membrane is gone and chromosomes are lined up in the middle of the cell
Describe Anaphase
Chromosomes are pulled to opposite ends of the cell by spindle fibres
Describe telophase
Two new nuclear membranes are formed around the two pairs of chromosomes
Describe cytokinesis
The cytoplasm is split forming two new identical daughter cells
Definitive
primary/sole treatment
Adjuvant
treatment given before the main treatment to improve the outcome
Neoadjuvant
Treatment given after the main treatment to reduce risk of recurrence of the disease
Salvage
Treatment given after the main treatments have failed to remove the residual disease
Dose intense regimes
higher doses of cytotoxic drugs per cycle while maintaining the standard cycle intervals
Dose dense regimes
Uses higher doses of cytotoxic drug doses while decreasing the interval between each cycle
Alkylating agents MOA (cyclophosphamide, ifosfamide)
attaches an alkyl group to DNA resulting in DNA strands breaking, mispairing, and sticking together resulting in DNA synthesis inhibition causing cell death
Platinum compounds MOA (cisplatin, carboplatin)
Binds to nitrogen atoms forming cross links (ties DNA parts together) preventing DNA synthesis causing cell death
CIsplatin main area of toxicity
Nephrotoxicity as it is secreted by renal tubules
Oxaliplatin main area of toxicity
sensory neuropathy
Antimetabolites 2 main MOA
Pretend to be chemical pieces called metabolites and cause cell death by:
Sneaking into DNA as they look nearly identical which results in DNA being unable to carry out synthesis causing cell death
Blocking important enzymes used in building DNA resulting in the prevention of DNA rather than having to directly damage DNA
Functions of metabolites
These help make:
DNA
Proteins
Cell membranes
Energy
What are the 2 main subtypes of antimetabolites?
Folic acid (VitB9) antagonist
Purine + Pyrimidine analogues
Folic acid (VitB9) antagonist MOA
Blocks the action of folic acid by sneaking into cells as it looks similar to folic acid
Blocks the enzyme dihydrofolate reductase (DHFR), which prevents the conversion of dihydrofolate into tetrahydrofolate which is essential for DNA synthesis
Therefore cells cannot undergo DNA synthesis
Where do antimetabolites act in the cell cycle?
S phase mainly
Folinic Acid Rescue (Leucovorin)
Special form of folate which is able to bypass the blocked enzyme, dihydrofolate reductase (DHFR),
from methotrexate allowing non cancerous cells to recover and synthesis by restoring folate in them
Purine analogues MOA
Mimics A or G purines which are inserted into DNA resulting in faulty DNA causing cell death
Pyrimidine analogues MOA
Mimics C, T, and U, blocking enzymes required to create thymidine resulting in DNA being incomplete causing cell death
Topoisomerase
Enzyme which is responsible for preventing supercoiling of DNA by cutting it where it is tangled then resealing it
Topoisomerase inhibitors MOA
prevents the enzyme topoisomerase from resealing DNA after it has been cut resulting in DNA damage then cell death
Topoisomerase I Inhibitors MOA
prevents resealing of single strand DNA cuts
Topoisomerase II Inhibitors MOA
prevents resealing of double strand DNA cuts
Key toxicity of anthracyclines
cardiotoxicity due to the release of free radicals which cause damage to the cellular membrane of cardiomyocytes
Anthracyclines MOA
Insert themselves between DNA bases and interfere with topoisomerase II
Key toxicity of epipodophyllotoxins
Bone marrow suppression/myelosuppression (low RBC, white bloods cells, platelets)
Epipodophyllotoxins MOA
Prevents Topoisomerase II from resealing DNA after its cut, causing DNA damage to accumulate causing cell death
Tubulin Inhibitors MOA
Inhibit the action of microtubules to prevent cell division
What phase do tubulin inhibitors act in?
M phase
Vinca Alkaloids MOA
Prevent microtubules from forming by preventing polymerisation of tubulin (protein which microtubules are made up of) resulting in cell division stopping in metaphase

Taxanes + Epothiolones MOA
Freezes microtubules preventing them from breaking down (normally the build then break down) resulting in dysfunctional mitotic spindles causing cell division to stop
Targeted Therapy
They act on specific molecular targets and signalling pathways associated with cancer cells. Only useful in pts who tumour has a specific gene mutation that codes for that target.
Small molecular drugs
small chemical compounds that are able to cross the cell membrane to enter cancer cells and target enzymes or DNA processes

Large molecular drugs
Large proteins that are unable to cross the cell membrane so they work by binding to targets on cancer cells to exert their effects

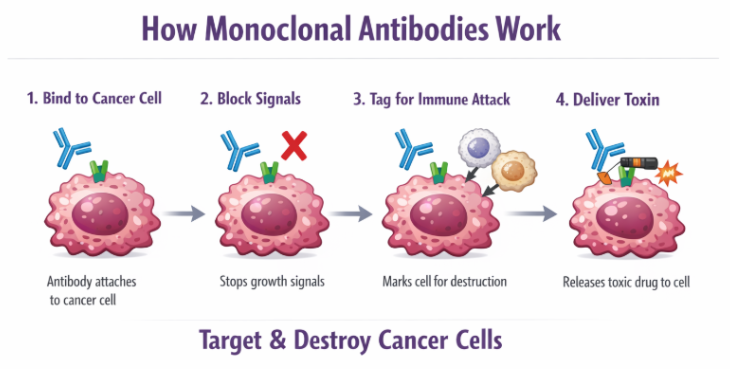
Monoclonal Antibodies MOA (Large molecular drugs)
Recognise and attach to one specific antigen on cancer cells
Block growth signals on the receptor of cancer cells preventing them from receiving growth signals
Marks the cancer cell for our immune system to attack and destroy the cell
Some antibodies carry the drug directly to the cancer cell which reduces damage to normal cells
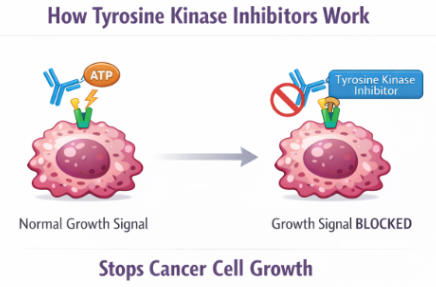
Tyrosine kinase Inhibitors (TKIs) MOA (Small molecular drugs)
TKIs enter the cell membrane
Bind to the ATP binding site of tyrosine kinase (enzyme which is overactive in cancer cells) preventing it from working
Cancer cells would use this ATP for energy to active growth signals but without it the signalling pathway is blocked
Cancer cells cannot undergo replication causing apoptosis
Angiogenesis Inhibitors MOA
Cancer cell releases vascular endothelial growth factor (VEGF) to stimulate vessel growth
Drug blocks VEGF or the receptor
VEGF is unable to activate the receptor on endothelial cells
New blood vessels cannot form to grow towards the tumour
Tumour becomes starved of oxygen and nutrients preventing tumour growth
MAPK Pathway (RAS-RAF-MEK-ERK)
Controls cell growth and proliferation
PI3K-AKT-mTOR Pathway
Controls cell survival (AKT a key protein that prevents cancer cells from cell programmed death), metabolism, and growth
Immune checkpoint inhibitors (ICIs)
Subset of monoclonal antibodies that BLOCK inhibitory pathways in T cells allowing our immune system to recognise and attack cancer cells
MOA of PD-1
Expressed on the surface of T cells and acts as a ‘off switch’ which can be used by healthy cells to downregulate T cells when they become overactive and start attacking healthy cells
How is PD-1 activated?
Normal cells will have a complementary molecule on their surface called PD-L1 which will bind with PD-1 on T cells causing them to ‘off’.
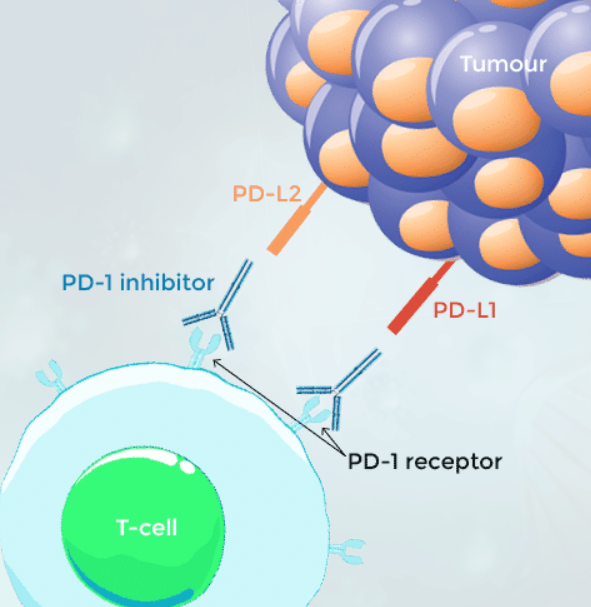
MOA of action of ICIs which target the PD-1 pathway
These drugs bind to PD-1 receptors on T cells preventing tumour cells with PD-L1 ligand from binding with it to ‘off’ its response.
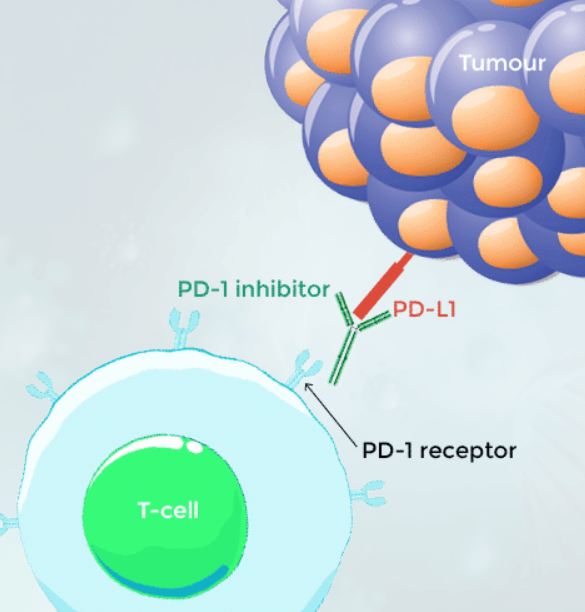
MOA of action of ICIs which target the PD-L1 pathway
These drugs bind to PD-L1 on tumour cells preventing binding of PD-L1 to PD-1.
What 2 signals must be received in order for T cells to be activated?
Antigen recognition: T cell receptor (TCR) binds antigen MHC
Co stimulation: CD28 on T cells binds to B7 (CD80/86 on antigen presenting cells) = T cell activation
CTLA-4 MOA
When CTLA-4 binds to B7 (CD80/86) it blocks CD28 stimulation which results in an inhibitory signal being sent to the T cell. This results in inhibition of T cell activity as CTLA-4 is responsible for stopping overactivation of the immune system.
CTLA-4 in cancer cells
Cancer cells exploit this pathway by promoting inappropriate CTLA-4 signalling to inhibit T cell activity, allowing them to survive and proliferate.
CTLA-4 inhibitors
Blocks CTLA-4 causing CD28 to bind to B7 allowing activation of T cells to recognise and attack cancer cells.
Hormonal therapy
mainly targets male and female hormones androgens and oestrogens as some cancer types are driven to grow by the presence of particular hormones
What are the 4 main classes of hormonal therapy?
Anti-androgens
Aromatase inhibitors
Gonadotropin-releasing hormone (GnRH) agonist
Selective Oestrogen Receptor Modulators (SERMs)
Hormonal therapy: Anti-androgens
Mainly block or inhibit androgens such as testosterone and dihydrotestosterone (DHT)
Anti androgen MOA: Inhibition of androgen synthesis
Reduces the amount of testosterone made by acting on the hypothalamic pituitary gonadal axis causing LH suppression. This suppression prevents synthesis of testosterone.
e.g. Abiraterone
Anti androgen MOA: Inhibition of 5a reductase
Prevents 5a reductase, an enzyme responsible for converting testosterone to DHT, resulting in low DHT levels which is helpful in preventing prostate growth
e.g. Finasteride
Anti androgen MOA: Androgen receptor blockade
Directly blocks androgen receptors which stops testosterone and DHT from binding to these receptors. Therefore, cells do not respond to them causing a reduction in male characteristics such as less hair and decrease prostate growth
e.g Bicalutamide, spironolactone
Anti androgen MOA: Suppression of androgen release
Suppression of LH secretion which results in medical castration due to decreased testosterone synthesis
Hormonal therapy: Aromatase Inhibitors
Targets aromatase which is an enzyme responsible for converting androgens into oestrogen. Prevention of this convertion results in decreased levels of oestrogen
Hormonal therapy: 2 types of Aromatase inhibitors
Non-steroidal (reversible inhibitors: Binds temporarily to aromatase to inhibit its enzyme activity
E.g. letrozole
Steroidal (irreversible inhibitors): Acts as a false substrate (molecule an enzyme acts on which gets converted to a product) and permanently inactivates aromatase
E.g. Exemestane
Hormonal therapy: Gonadotropin-releasing hormone agonist (GnRH)
Mimics the natural GnRH but the effect depends on how the drug is administered:
GnRH agonist: Initial stimulation (Flare effect)
When first given they stimulate GnRH receptors causing a temporary increase in testosterone and oestrogen

GnRH agonist: Continuous use = Suppression
Continuous administration results in GnRH receptors becoming desensitised and downregulated causing a decrease in testosterone and oestrogen.
Hormonal Therapy: Selective Oestrogen Receptor Modulators (SERMs)
Binds to oestrogen receptors and act as either antagonist or agonist depending on the tissue:
Antagonists: Blocks oestrogen effects in specific tissues
Agonist: mimics oestrogen in specific tissues

Pharmacodynamics (PD)
What the DRUG does to the body
PD Interactions
Synergistic Interactions: Drugs which are combined produce a greater effect, resulting in increased anti-cancer activity
Additive Interactions: Combination of drugs results in increased toxicity
Antagonistic Interactions: Combination of drugs results in reduced effectiveness
Pharmacokinetics (PK)
What the BODY does the drug
PD (ADME)
Absorption: Movement of the drug from the site of administration into the bloodstream and is affected by the route:
Oral, IV, IM
Distribution: Movement of the drug from blood into tissues and is affected by:
Protein binding
Tissue perfusion
Lipid solubility
Metabolism:
Modification of the drug mainly in the liver
Cytochrome P450 (CYP450) which are enzymes in the liver responsible for metabolising >90% of drugs
Excretion/elimination:
Removal of drugs from the body via the kidneys mainly