mol gen exam 2 (DENSE)
1/231
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
232 Terms
true/false: mutation rates are a phenotype
true
endonucleases act like what?
a sword — cuts straight down in one spot
exonucleases act like what?
pacman — chews along the line, starting from the ends or a nick
BRCA-2: what is it? what kind of DNA repair system does it affect when damaged? consequences?
a gene that is significant to inherited defects in DNA repair — affects repair by homologous recombination, leading to breast/ovarian cancers
mismatch repair: what does it do? what enzymes does it use?
mechanism used by all cells to repair many of the errors that escape repair by DNA polymerase
Mut proteins (MutH, MutL, MutS)
nucleotide excision repair: what does it repair? what is the genetic condition where this is nonfunctioning?
repairs pyrimidine dimers (sun damage!) to prevent skin cancers
xerodosa pigmentosum — they can’t be in the sun at all or it’ll do remarkable damage to their genome, leading to other cancers
what is the standard error rate in e coli:
before proofreading
with proofreading
with mismatch repair (MMR)
1 in 10^5
1 in 10^7
1 in 10^9
improves by 2 orders of magnitude (100x) each time — proofreading is 100x better than none, and MMR is 100x than proofreading alone.
which strand, by definition, will contain the incorrect nucleotide in the event of a missense mutation?
the newly synthesized strand
explain strand identification — overview
for MMR, we have to be able to identify the new strand from the template strand. consider three systems: A allows for strand ID, system B repairs lesions randomly, system C has no MMR at all.
obviously A is the best, but B and C are actually equally bad: repairing lesions randomly is no different than having no repair system at all

use this image for diagramming systems B and C in the following questions
assume the T is the correct nucleotide, and the G opposite it was incorrectly incorporated.
.
strand ID — system C (no MMR) drawing + results
the gene is not repaired at all. because both are separately used as templates, one correct and one incorrect, replication of this gene will result in ½ of the newly synthesized DNA being correct.

strand ID — system B (MMR, but no strand ID) drawing + results
because the system can’t identify which is the “correct” strand, it’ll randomly choose which one to act on… half of the time it will repair the mismatch; half of the time it will fix the mismatch (MAKE PERMANENT). this also results in ½ of the resulting genes being correct

is it worth having MMR at all if you can’t recognize the newly synthesized strand?
no, it makes no difference.
what is the difference between mut and Mut? what repair system are they involved in?
mut are genes; Mut are the proteins they code for
MMR
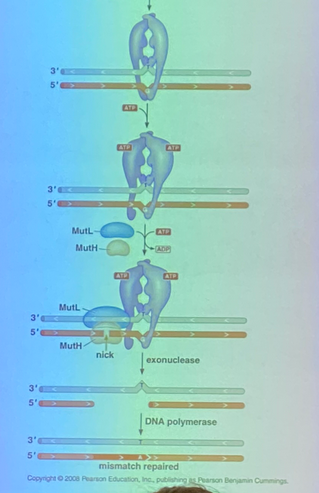
mutator proteins: what are their names and collectively what 3 things do they have to do?
MutS, MutH, MutL — mutator proteins that all do different things for MMR, but have to identify the mismatch/lesion, have to identify newly synthesized strand from the template, and have to start the process of repair.
MutS: what does it interact with + where + when? what’s special about it? mechanism of action (3)?
interacts with the replisome at the place where mismatches are occurring (right place, right time to replace mismatches)
is a shape changing protein and have multiple conformations
recognizes mismatches in major groove, changes conformation, hydrolyzes ATP and twists the helix to form a kink where MutH and MutL can come in
MutH: describe the specific way that it binds to DNA. is MutH an endo or exonuclease? 4 steps of repair after binding?
binds DNA in such a way it is directed to the unmethylated strand (VERY FRESH newly synthesized strand) in hemimethylated DNA
an endonuclease
will nick the unmethylated strand a bit further down (due to the geometry of the complex). a helicase comes in and unwinds things, and an exonuclease chews back the nick → removes the mismatched base that was recognized by MutS AND all of the bases leading up to it. now we need DNA pol II and ligase to fill in the gap
why don’t Mut proteins don’t work well in areas of DNA that are not freshly synthesized?
because MutH has to be directed + bound to the unmethylated strand of very freshly synthesized DNA (exists only during the cooldown period between replication events).
MutL — function?
a linker protein — holds MutH and MutS together in this complex
(also attracts the helicase to come in, but this wasn’t mentioned to most of the class)
mismatch repair is a solution to what three problems?
how do we identify the mismatch/lesion?
how do we recognize the newly synthesized (problematic) strand?
how do we repair the incorrect nucleotide?
MMR solution to question 1: how do we identify the mismatch/lesion?
MutS looks at arrangement of atoms in the major groove — a mismatch looks different than the normal watson-crick base pairs. once bound, will change conformation using ATP and form a kink in the helix
how is recognition of a mismatch in MMR coupled with replication?
MutS interacts with sliding clamp of replisome — falls off occasionally, and good evidence shows that it falls off when it encounters these sorts of regions — likely how we recognize it
MMR solution to question 2: how do we recognize the newly synthesized (problematic) strand?
the freshly new strand is not methylated, which is recognized by MutH. MutH is then directed to the lesion on the unmethylated strand thanks to MutL (linking protein) after the DNA is kinked by MutS!
MMR solution to question 3: how do we repair the incorrect nucleotide? (3 steps)
MutH (endonuclease) nicks the template strand a little ways up/downstream (the process works regardless of direction).
an exonuclease (pacman) comes in to chew away towards the mismatch — there are two kinds, the kind depends on the direction it needs to chew; we don’t need to know the name of either.
DNA pol III synthesizes through the gap, but can’t mend the gap as per usual, so ligase comes in and glues it.
we and gram positive bacteria don’t methylate our DNA in the same way, yet we both do MMR, meaning we must have slightly different mechanisms of MMR.
tbh, ask others for their notes when you meet for group problem set. not sure if i got all of the details or context down right
during lagging strand synthesis, there is a gap in time between when DNA pol I removes/replaces RNA primers and ligation, so these little “nicks” exist there— in eukaryotic DNA polymerization, at least during lagging strand synthesis, the machinery responsible for recognizing the new strand can do so by recognizing the presence of those nicks. thus, we probably can’t use the same mechanism during leading strand synthesis… therefore we can expect MMR to work better during lagging strand synthesis.
true or false: the lagging strand mutation rates are significantly lower than those of the leading strand
false: the mutation rates are so low, it’s hard to generate reliable data to compare rates between leading and lagging strand synthesis. no reports we have that do suggest this are confident enough to claim definitively
HOWEVER, it is clear that we have MMR during synthesis for both strands.
what proteins do euks have for MMR?
MutS homolog and MutL homolog, NO MutH!
true/false: eukaryotes and prokaryotes have MutH
false. euks do not have MutH, but we do have an enzyme that nicks DNA on the newly synthesized strand… and that enzyme is MutL!
idea of how MutL works in euks
MutL randomly nicks during synthesis, but those nicks are far more commonly made on the newly synthesized strand than the template strand, and that is responsible for repair because once we have the nick, we can bring in an exonuclease that can somehow recognize the MutS/MutL complex that is bound at the site of these lesions. the nuclease will chew back the lesion and a region of neighboring newly synthesized DNA so that we can fill in the gap using DNA pol and ligase.
since MutL nicking the DNA is somewhat at random (occasionally nicking the template strand instead), is it still worth having DNA repair?
yes — so long as it nicks the newly synthesized strand more often, it’s better than having nothing.
does DNA pol ever add RNA nucleotides?
yes, somewhere between 1 in 5k-10k nucleotides — considering there are 6 billion nucleotides in your genome, so when we copy the genome, you’re gonna have a lot of RNA.
do the RNA nucleotides persist in your genome?
no — ribonucleotide excision repair gets rid of them.
how does ribonucleotide excision repair occur?
it recognizes all of the places in the genome where you add RNA nucleotides instead of DNA nucleotides. first step is utilization of RNaseR— an endonuclease that cleaves @ junction between DNA and RNA nucleotides on the newly synthesized strand and not the template.
**ARE THERE OTHER STEPS??
happens during both leading and lagging strand synthesis
if one of the jobs of MutL is to nick the genome in a way that helps the cell identify the newly identified strand from the template, couldn’t it be that RER directs MMR in eukaryotic cells to lesions to repair? explain using the experiment using yeast
got rid of RNaseR so that the RNA-DNA lesions wouldn’t be repaired correctly
turns out that rate of MMR also got worse, to the point where we might as well not have MMR at ALL!
suggests that the nicking that occurs in recognizing improperly added RNA nucleotides allows us to distinguish the newly synthesized strand in DNA in a way that we can direct MMR, at least in part, to the correct strand to be repaired.
MMR corrects errors of ___. moving forward, will the repair mechanisms discussed correct the same thing?
synthesis — errors due to improperly added nucleotides from polymerase
no — errors due to ENVIRONMENTAL DAMAGE!
do mechanisms for repairing environmental damage require strand ID? why/why not?
no — damage can occur on either strand and anywhere in the genome
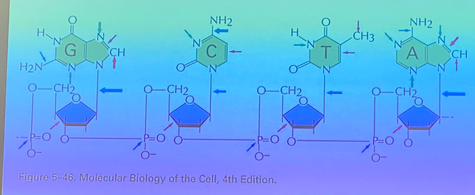
EXTREMELY IMPORTANT IMAGE showing all of the breakable covalent bonds that we find in DNA
arrow colors show different reaction types that can break these bonds
arrow SIZE shows FREQUENCY of that splice occurring.
turn this card back around and look at that image some more.
what is the most frequent covalent bond spliced in DNA?
the bond linking the sugar to the nitrogen base → the base just falls off, even in double stranded form.
especially in PURINES — DEPURINATION! occurs ~5000x per cell per day in your organism. compare to depyramidization — only 50x per cell per day
what is another common covalent bond break we find in DNA, other than depurination/depyramidization? what nucleotides are prone to this?
deamination — G and C and A all have the potential to be deaminated just because there is water everywhere and they’re reactive
consequences of deamination
C is deaminated to U ~100x per cell per day
A is deaminated to hypoxanthine; G is deaminated to xanthine. both at lower rate than C is deaminated, but still significantly.
if not repaired, leads to permanent base substitution mutation — draw the replication events!
true/false: regardless of what nucleotide is deaminated/depurinated/depyrimidized, we can build a machine that recognizes all of the incorrect pairs and get rid of it
false. we can get rid of all U, hypoxanthine, and xanthine in DNA, but deamination of 5-methyl cytosine becomes THYMINE! obviously we can’t build something that cuts out all Ts in our DNA.
base excision repair mechanism (BER)
essentially one mechanism by which all deaminations of C/A/G (U/hypoxanthine/xanthine) are repaired
if we see a deaminated C into U, what base is likely across from it?
G — because it was originally a regular C that later lost its amine!
fixing spontaneous conversion of adenine to hypoxanthine, guanine to xanthine, cytosine to uracil — what do we need to get started?
first we need a set of enzymes that will recognize the presence of hypoxanthine, xanthine, and U in DNA. there are multiple versions of each — glycosyltransferases!
first family of proteins involved in BER
glycosyltransferases
describe glycosyltransferases (4)
recognize bad base pairs, like RNA-DNA lesions (xanthine, hypoxanthine, uracil). some can even recognize G-T base pairs
can exchange/transfer a glycosidic bond (connects the sugar to the base)
once the bad pair is recognized, they are flipped out of the double helix and their chemical reactivities are triggered — the base is cut off. looks like depurination/depyrimidation, but done by an enzyme instead of water
result: looks like the base fell off by itself and leaves a gap/lesion!
how are gaps left behind by glycosyltransferases repaired: give a quick overview, name what enzyme(s) you need + if they has endonuclease or exonuclease activity, and explain how it works in a liiittle more detail
basically: cut out the backbone, use the existing base as a template, and fill the gap.
need an endonuclease (sword). acts only @ site at which nucleotides had been depurinated/depyrimidated. called the AP (apurinated/apyrimidated) endonuclease/exonuclease — it’s BOTH!
the AP endonuclease/exonuclease cuts like a sword at a phosphodiester linkage, then pacman-chews out the nucleotide for which there’s no base. pol I does its thing, then ligase glues it in.
true/false: from this point forward, the pathways for fixing deamination and depurination are the same
true
nucleotide excision repair (NER) deals with what kind of damage?
deals with chemical modifications thats occur to your genome as a result of environmental exposure — damage that results in the formation of covalent bonds that disrupt the ability to copy + express your genome the right way.
thymine dimers — how do they form?
DNA absorbs UV really well, causing electrons to get excited → covalent bonds get made and broken.
more broadly referred to as pyrimidine dimers, as they can form from any pyrimidine (C-C, C-T, T-C)
consequences of pyrimidine dimers
the same as for the other sorts of lesions that will be repaired by NER — this portion of DNA can’t be replicated, transcribed, etc
what are bulky adducts and where do they most commonly form?
reactive organic molecules that can covalently link to your DNA. our cells are FILLED with them!
most commonly linked at reactive sites on nitrogenous bases
similar concept to smth like a peanut allergy, in which a bulky adduct binds covalently to your proteins instead of to DNA, but same idea
direct correlation between how carcinogenic a species is and how likely it is to form adducts with your DNA, i.e. smoking!
end result of bulky adducts and pyrimidine dimers
when DNA pols get to these species, they have no template strand because whatever modification has occurred will interrupt the template’s ability to form the H bonds it should be making
also disrupts the helix as it’s made less flexible
→→ polymerases are inhibited in several ways at once!
draw a thymine dimer
what happens when polymerase reaches the dimer? what happens if this DNA goes on to another round of replication?
it stalls + can’t use these thymines as a template → resulting DNA will have a gap that is likely several nucleotides in length! if 2+ are missing, we especially have a problem bc we don’t have a pol that can read the template there.
if it goes to another round of replication, we get one complete strand and one that has a gap on BOTH strands → GENOMIC INSTABILITY → DRAMATIC ISSUES
the pathway thru which we deal with both pyrimidine dimers and bulky adducts — name the process and enzymes
NER
Uvr — 4 types. UvrA, B, C, and D.
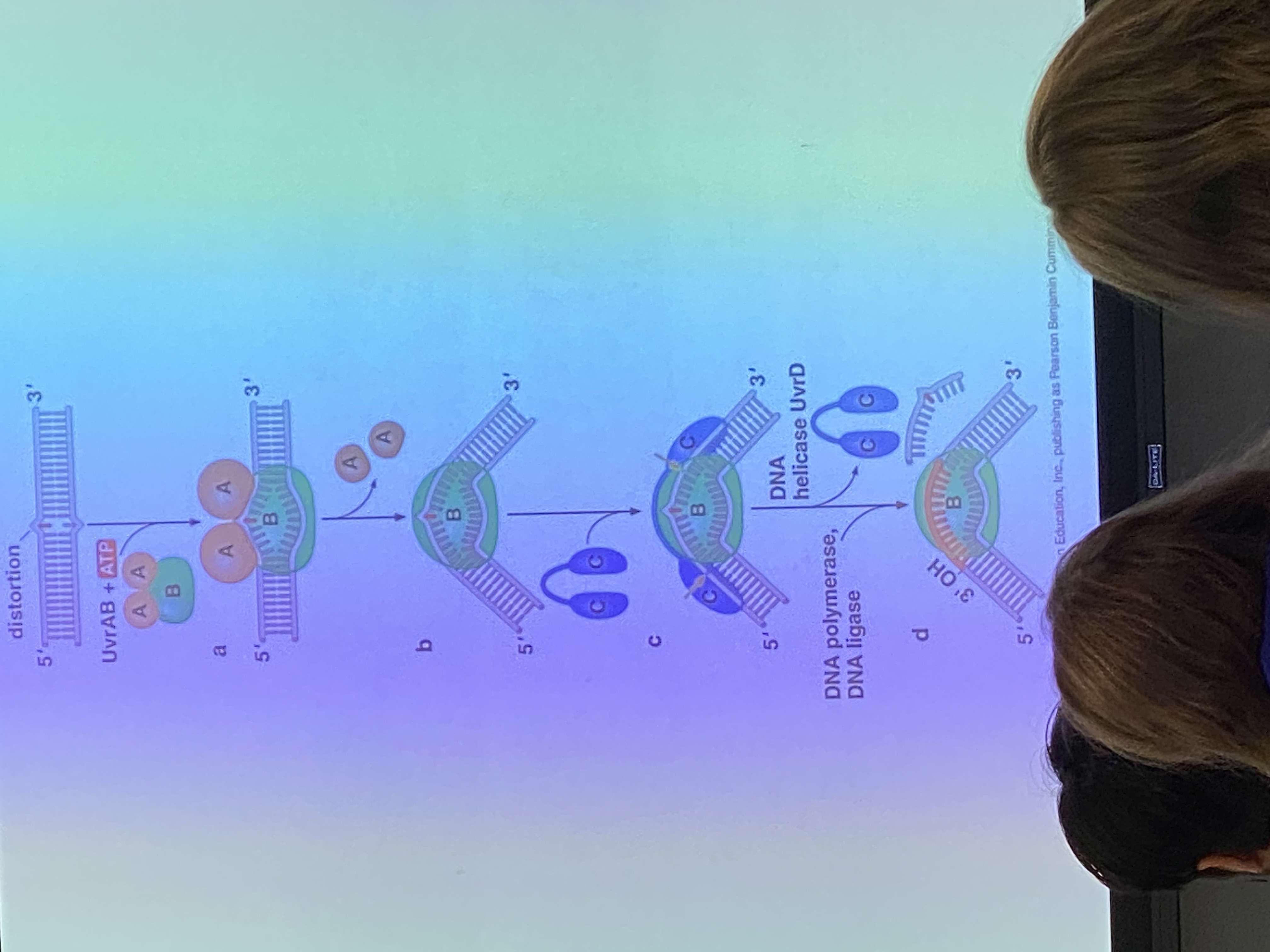
Uvr A/B do what? (2)
recognition — A recognizes the distortion that occurs in the phosphodiester backbone and brings B to the lesion site.
once the recognition site sits down here, A is kicked out and exchanged for UvrC
UvrB/C do what?
what does the strand look like as a result?
what do we do to totally get rid of the damaged portion?
endonucleases (swords). very precise. B cuts 8 nucleotides on one side of the lesion, C cuts 12 in opposite direction.
we’ve clipped the strand on which the thymine dimer is present so it can be removed — the phosphodiester linkage is interrupted in 2 spots. what’s holding that strand on to its complementary strand? — stacking interactions and H bonds.
unwind the helix and it’ll basically fall off!
what does UvrD do? what are we left with?
it is a helicase! we want to unwind the DNA to make the clipped-out strand dissociate from the complementary strand
we are left with a giant gap of 20 nucleotides that can be used as templates + recognized by pol 1 to fill in the gap, ligase glues back together!
true/false: our ability to perform NER (UvrA/B/C/D) is always somewhat constant
false. “therapeutic response” — we get better with repair the more exposure we have to sun (not to say you SHOULD get a ton more sun exposure)
if there is ZERO UV exposure, there is ZERO UV nucleotide excision repair! → xerodosa pigmentosum
other way to deal with the issue of pyrimidine dimers and bulky adducts, other than NER
translesion DNA polymerases
how do translesion DNA polymerases work?
when it encounters a dimer, the nucleotides that it adds are “random” (N=any of the 4 nucleotides!) — NOT in watson-crick base pairs!
if you rely on these for dealing with dimers, by definition they are mutagenic… but we use them anyway! thus there must be a cost/benefit analysis that makes this process better than just having these bases entirely missing on both strands. ultimately, this mutagenic result is bad, but the alternative is HORRIBLE, so we take what we can get!

true/false: both NER and translesion DNA polymerases work all the time and work in such a way that the problem is solved by both mechanisms
true
do BER and NER repair mismatches
NO — we define mismatches as when polymerase adds the wrong nucleotides
instead, they solve two different sets of problems, neither of which is linked directly to DNA synthesis, neither of which needs to recognize the newly synthesized strand
BER solves deamination, depurination, and depyrimidation. enzymes involved: glycosyltransferases, AP endo/exonucleases
NER solves pyrimidine dimerization and buildup of bulky adducts in DNA — major problem bc DNA doesn’t get copied. enzymes involved: Uvr proteins A/B/C/D.
nucleotide excision repair machines actually associate with RNA polymerase (i think that’s whay he said? recording is muffled)— transcription-coupled repair. this works best where?
in regions of genome that are being actively expressed
what two mechanisms are employed to deal with the potential failure to copy the genome in its entirety?
NER
translesion DNA polymerases that can copy in a way that is independent of the templates they use — add a randomized selection of nucleotides
most common reason we get double stranded breaks:
we constantly get nicks in DNA, sometimes in part bc we’re exposed to ionizing radiation like x rays, but mostly because there’s water everywhere and those bonds just hydrolyze
ligases famously deal with this problem, but the ability to repair is never perfect. occasionally (12-20x per round of replication), DNA pol comes up against the nick. if not repaired by the time DNA pol arrives, then we see that one resulting DNA molecule is fine while the other is missing a chunk from the middle
problem bc it does things like remove regions of chromosomes from their centromeres, or remove telomeres → mitosis gets mucked up when DNA fragments get separated from centrosomes and telomeres
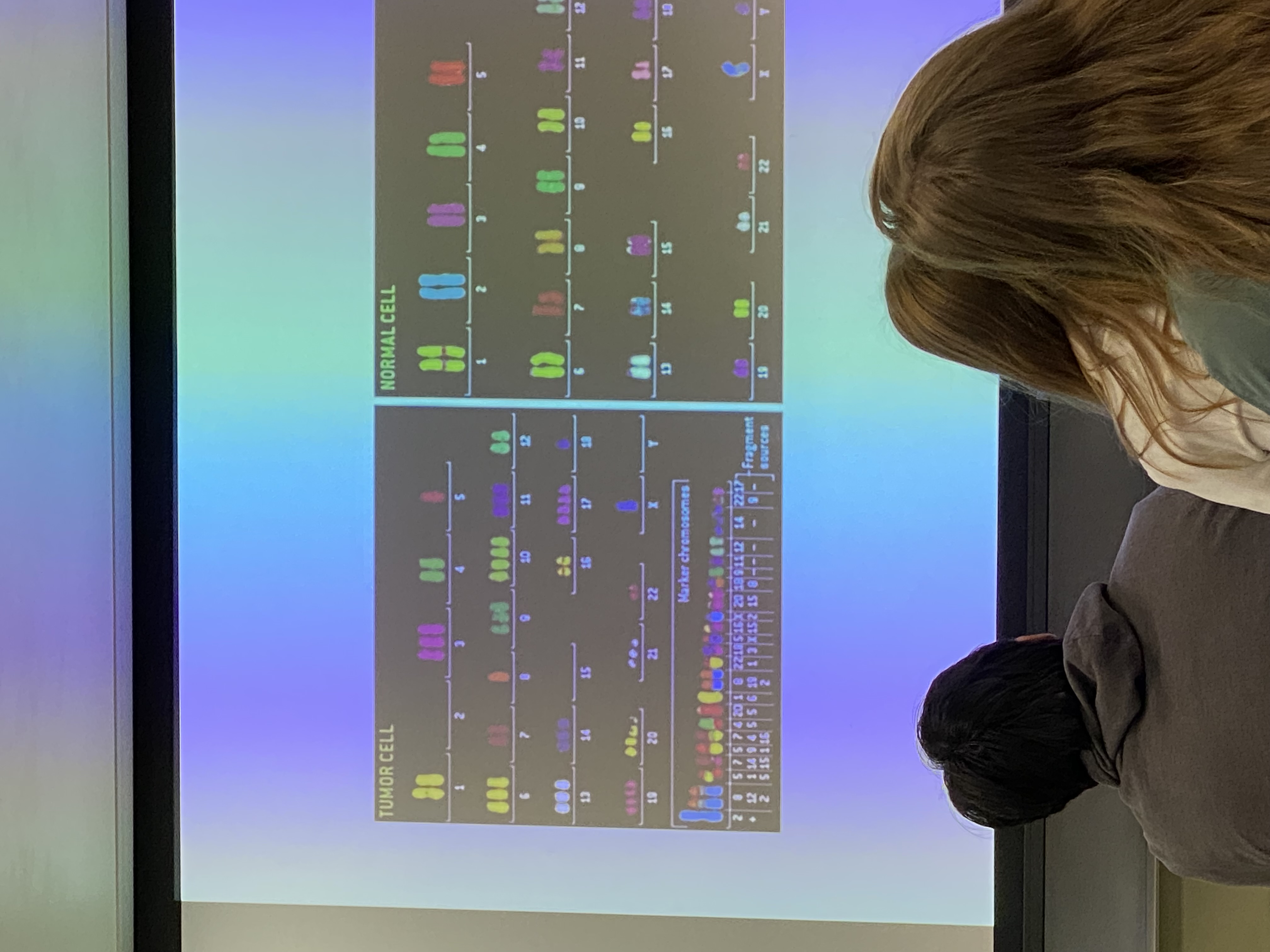
note the marker chromosomes at the bottom of diagram for tumor cell: fusions/chimeric chromosomes. famous example is the Philadelphia chromosome/Kareemosome (lol)
chimeric chromosomes happen when?
when homologous recombination w DS break repair doesn’t work in the most effective/efficient way. we have machines to reduce likelihood that these will happen
two big and broad mechanisms that we understand best for repairing an accidental break
nonhomologous end-joining (NHEJ)
homologous recombination (HR)
NHEJ is inherently __?
mutagenic
HR: the info present @ site of break is _?
copied in using an unbroken copy of the genome
how does homologous recombination work?
identify region missing
use unbroken version of chromsome to copy that info and insert into the genome
CRISPR/Cas9 is an example of which: NHEJ or HR?
NHEJ if deleting a gene
homologous recombination if changing/replacing a gene
do humans do HR, NHEJ, both, or neither?
both — but by far, for the majority of our DS breaks, we repair them via NHEJ
(tend to do HR during DNA replication and during G2; CROSSING OVER!)
do e. coli do HR, NHEJ, both, or neither?
homologous recombination ONLY

accidental break diagram
accidental break diagram

image for NHEJ overview diagram
.
NHEJ: what happens first?
**image: drawing of DNA sections A, B, C with Ku proteins attached
Ku proteins bind around end of these DNA molecules @ their breaks and remove some (not many— less than 10 usually) nucleotides. EXOnucleases. → remove some of section B. activity of Ku does NOT guarantee that the ends of these DNA molecules will be blunt — make sticky cuts instead
Ku proteins bind at the broken ends of the DNA and chew back (exonuclease) some of section B. produce sticky, not blunt, cuts
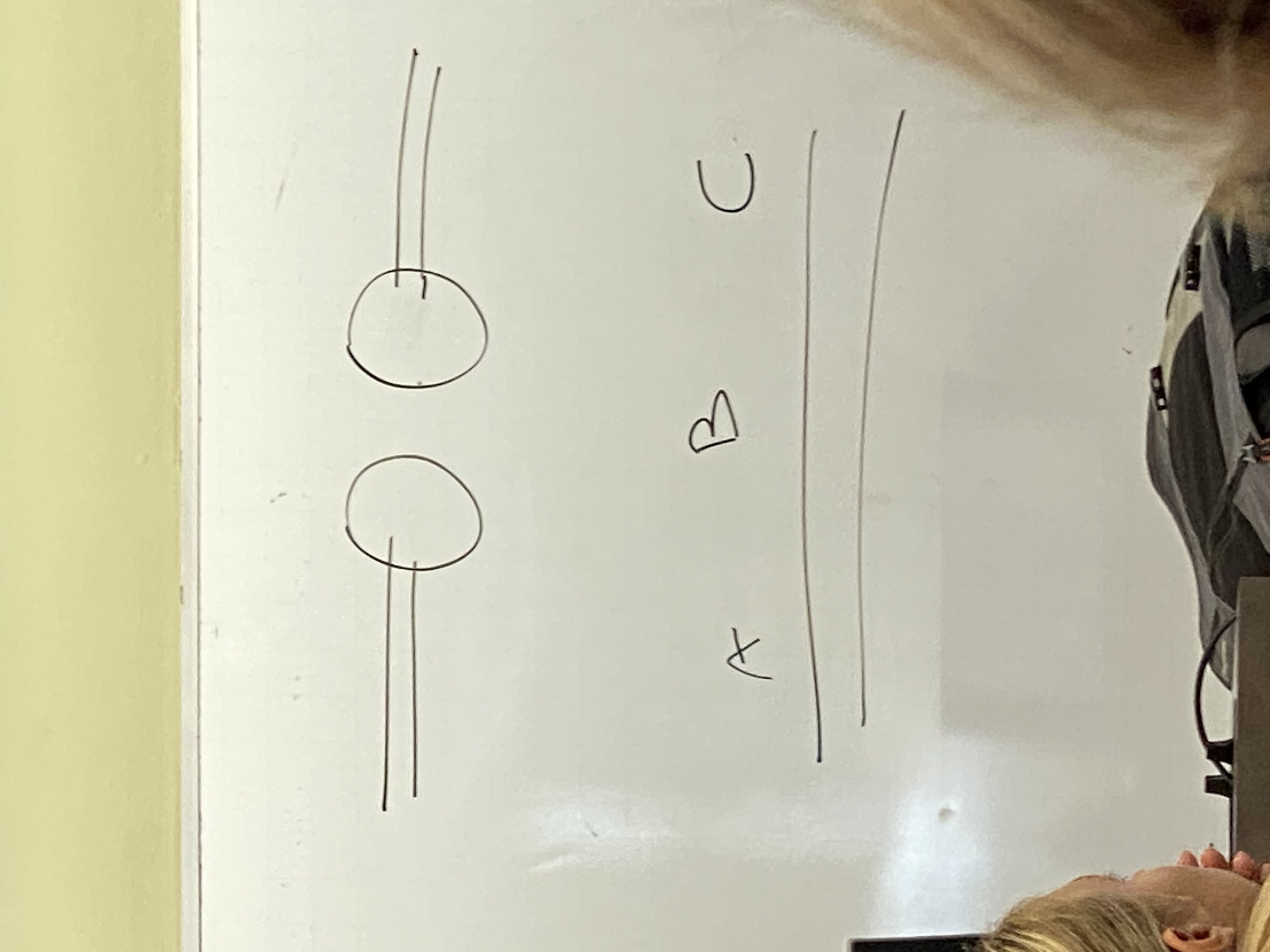
what is a blunt end on DNA? what is a sticky cut?
blunt: we have two molecules where we mirror base pairs on both strands, but the phosphodiester linkages broke at the same position on both strands.
sticky: broke at different positions on either strand — have overhangs. produced by Ku proteins. have little flags of SS DNA floating around.

need another protein to take the sticky ends and blunt them — name it
artemis
needed bc DNA ligase is capable of stitching these back together only if these ends are blunt
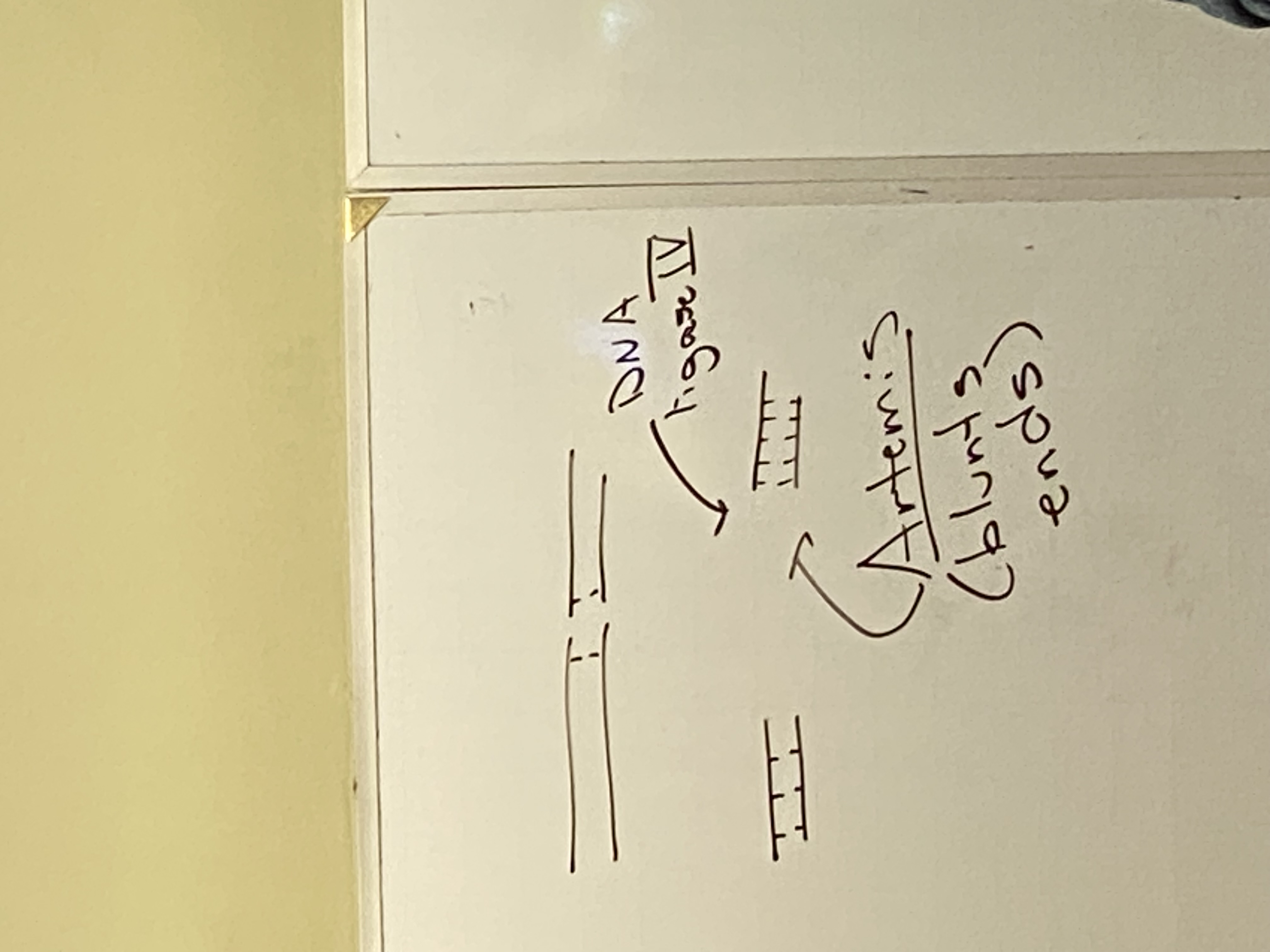
last protein in NHEJ is DNA ligase 4: result?
makes a DS DNA molecule that has sections A and C, but is lacking information from B
(top right corner of image)
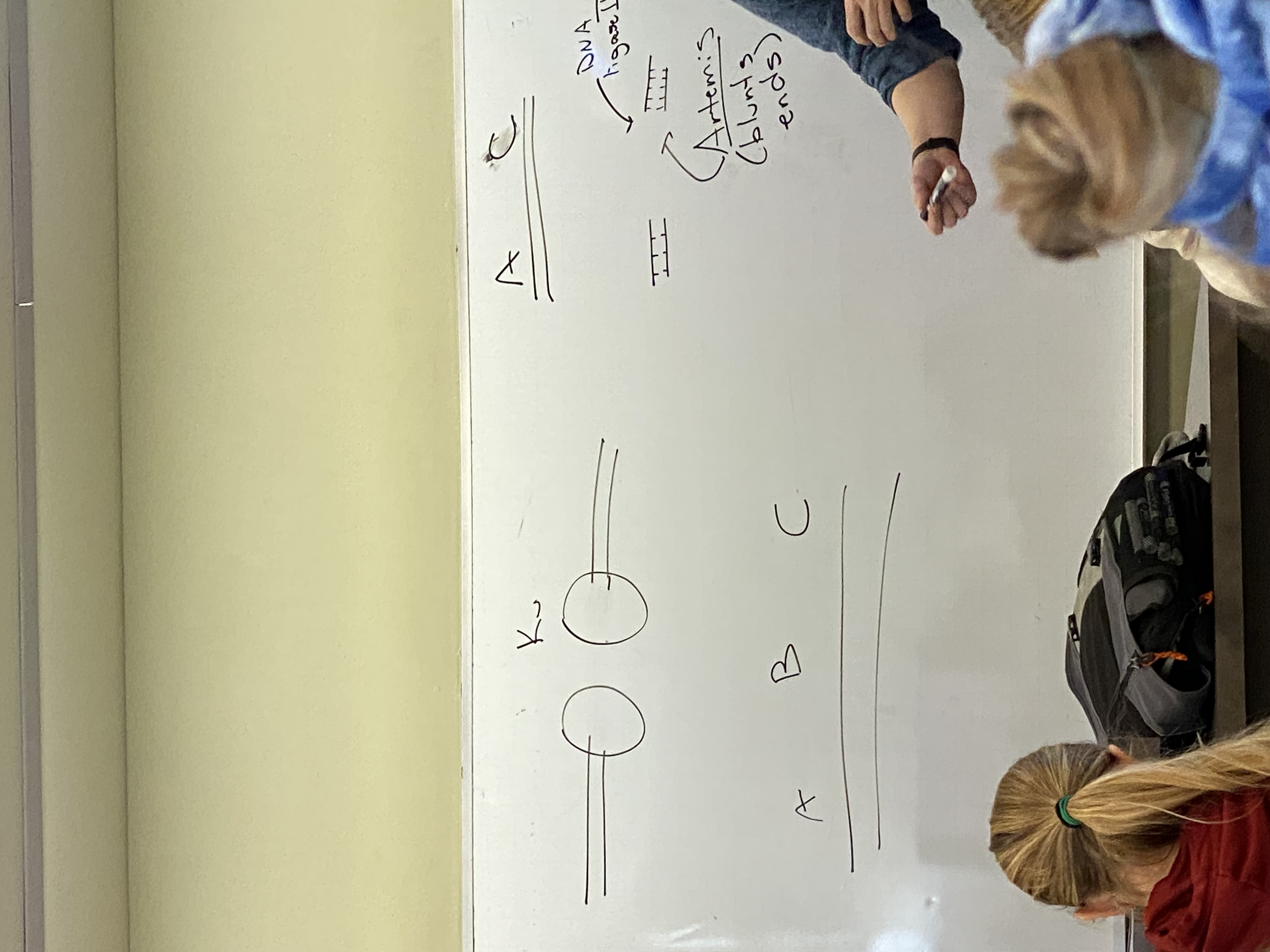
process of NHEJ takes how long in our cells?
~12 minutes
true/false: any time we have a double stranded break, there is going to be missing information
true… but remember that while NHEJ results in a loss of nucleotides, HR does not because we use the undamaged chromosome to fill in any gaps we had!
HR overview
using a second copy of the chromosome to fill in the missing information from a double stranded break
HR process — the “easy” part
we have a double stranded break on one chromosome, and an undamaged homologous chromosome. we will produce 3’ end overhangs in the middle of the damaged portion. we are now missing information from the middle (B).
form a bubble in the homolog to open it up and unwind it. build a machine that not only unwinds the homolog, but allows us to search for the particular sequence in the homolog that is complementary to the overhang. the overhang actually invades the bubble and searches for regions of complementarity. when it finds it, it pauses and the complex becomes relatively stable — called “strand invasion”
we have a free 3’ end and a template strand → we can polymerize DNA! ligase them together
top strand is easiest to understand— synthesis just continues until we reach the other end of the break, then they’re ligated together. same concept for the invading strand though — it’ll just cross back (forming a second Holliday junction) and ligate with the other end of that strand’s break. we have now replaced all of the missing info! next step: separate the chromosomes
true/false: we cannot form a Holliday junction without leaving some nucleotides unpaired where they cross
false: it appears that way because of how we have to draw it, but in reality it’s a super condensed structure, and we can form a Holliday junction with ZERO unpaired nucleotides
after reconciliation in HR, what information are we missing?
nothing! we’ve filled in the gap completely using the homolog as a template!
how to separate the chromosomes when they are linked together in HR?
the process is called resolution. requires cutting strands at both of the Holliday junctions. we have to cleave strands and ligate them back together to resolve one chromosome from the next
focus on this happening right where the strands cross when we cleave + ligate them back together.
one way to resolve: basically form two trapezoids— top resolves with top, bottom resolves with bottom. this way can be drawn; the other cannot
is resolution of the holliday junctions random, or is one kind of resolution more common?
random — we get each of the two possible products half of the time
possible products of HR
if cleaved + resolved in same way: parallel. patch recombination (non-crossing over) results. like a bandaid or skin graft
if cleaved + resolved in opposite ways: perpendicular. splice recombination (crossing over) results. like having a new hand sewn on
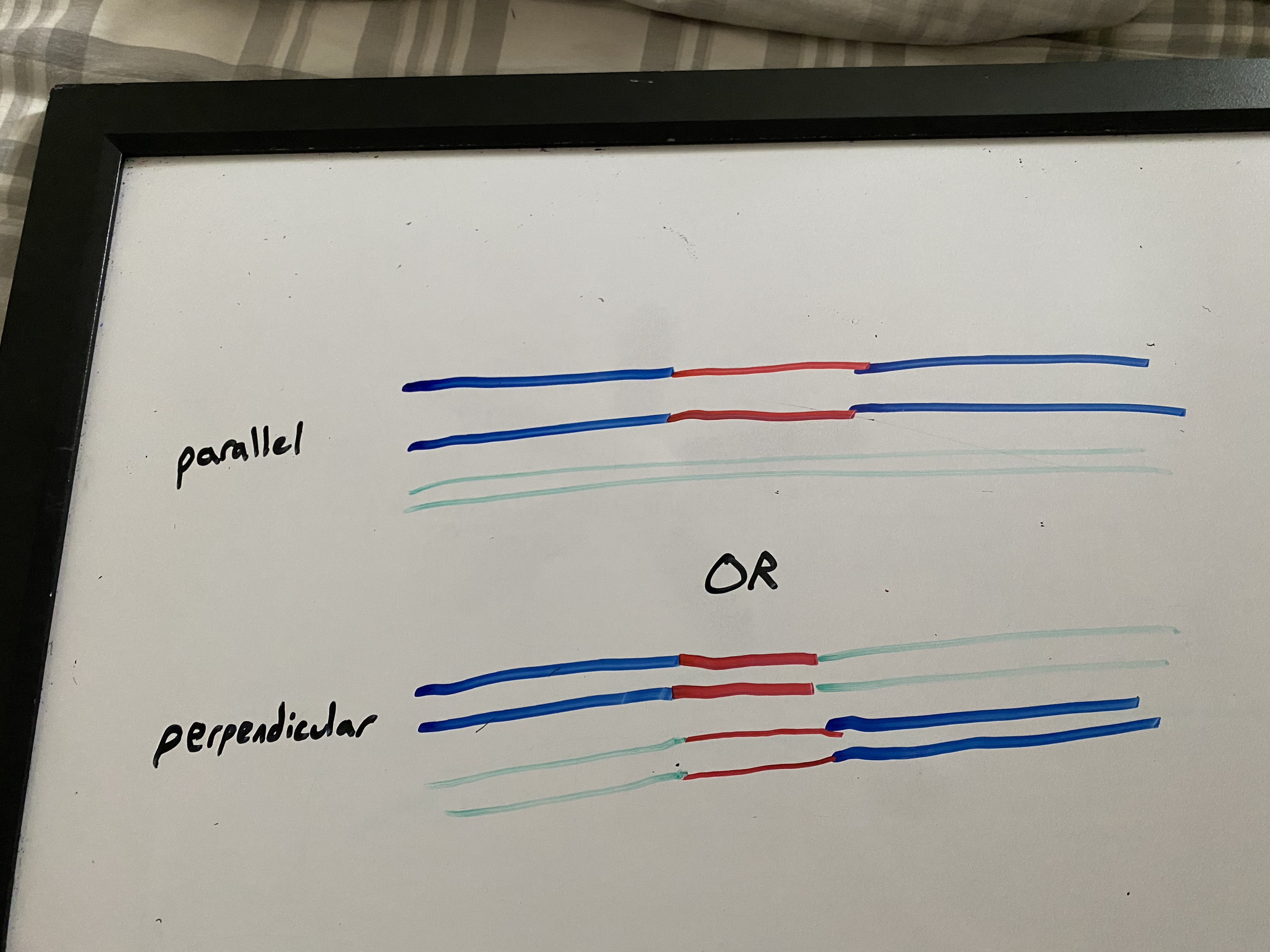
what is gene conversion?
in HR, we have 2 copies of each chromosome, which can swap info from one to another. thus, this does not guarantee that filled in info will be identical!
3 ways we might get a double stranded break in DNA
X ray radiation
DNA polymerase fell off during replication
during meiosis I, some enzyme created the DS break to initiate crossing over
HR: RecBCD and RecA pathway
purpose of BCD? activity on DS break? what does RecA do?
Rec BCD purpose: process the ends of the DS break to form the 3' overhangs.
goes along DNA, unwinding and cleaving DNA (this DNA is destroyed). when it hits a chi site (crossover hotspot instigator), the nuclease activity from RecD on the 3' end is halted. the DNA is no longer being cleaved as it is unwound on that strand -- the 5' continues getting chewed away, leaving this shorter than the 3' end -- overhang!
RecBCD directs RecA to the tail, which covers the tail with its hundreds of subunits. this complex of SS DNA and Rec A search the homologous chromosome for homology and form the stable complex -- strand invasion. DNA pol and ligase fill the gaps
HR: what are UvrA and UvrB?
UvrA is a protein that recognizes the structure of the holliday junction and brings in 2 UvrBs, which are ATPases -- provide the energy to drive the exchange of base pairs during branch migration (the junction MOVES along the DNA!)
for each holliday junction, how many possible cleavage sites are there for resolution? describe them
consists of the two DNA strands that have not crossed over
consists of the two DNA strands that HAVE crossed over
if both junctions cleaved in same way, patch recombination (skin graft; non crossing over). i.e. both at site 1, both at site 2
if both junctions cleaved in different ways, splice recombination results (new hand; crossing over). i.e. at 1 and 2, or at 2 and 1
for crossing over to occur, what must happen first? what enzyme does it use?
we must clip the DNA using enzyme Spo11 (in euks), which binds to the chromosome and semi-randomly clips them in prophase I of meiosis I
do prokaryotes, eukaryotes, or both do crossing over?
euks only — it is a DNA repair mechanism that we have co-opted for genetic diversity
how many crossing over events are typical for each chromosome?
the bigger the chromosome, the more crossing over events there will be
in big ones, 8-10 crossing over events per pair, so 16-20 recombination in total (remember that not all recombination is crossing over; some of these are just the patches that don’t result in gene swapping).
what is the buffer zone in crossing over?
a little safety bubble when crossing over occurs in one spot, so that other crossing over events can’t happen too close
if nonfunctional, leads to harlequin patterning where an incredible amount of crossing over occurs in a small space!