BMM Module 8: Molecular Dynamics
1/13
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
14 Terms
What is MD
computer simulations that allow us to study atoms and molecules and their interaciton over a period of time using known laws of physics (newtonion)
example: study how cholesterol inserts into the membrane
why do we need MD?
bridge theory and experiment:
practical model when experiments are too expensive or difficult
But simulations are hard to prove to be correct
Capture atomic motions or interactions that cannot be observed in the lab
Fills the gap where other measurements fall short like NMR or FRET
examples
local/global conformations
enzyme substrate binding
free energy determination
protein folding
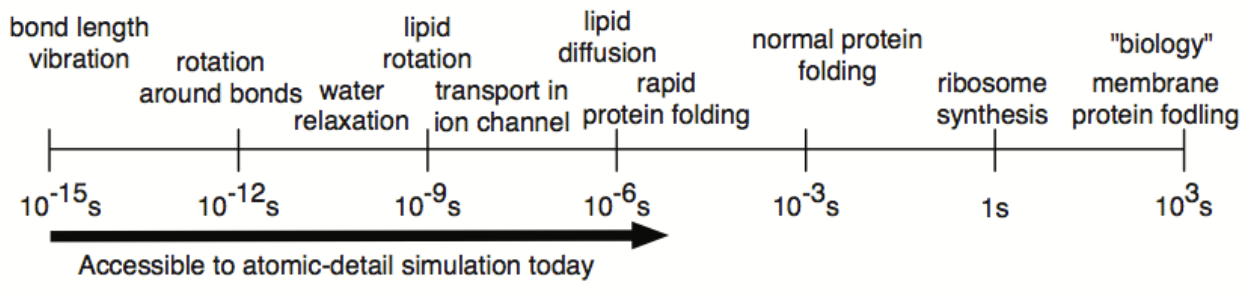
today MD simulations typically range from a 100’s of ns to a few µs
(normal protien folding is however in the range of ms)
How does MD work?
The force field determines the energy of the system: bonded + non bonded interactions
Non bonded interactions combine attraction and repulsion and therefore cause the biggest movement
EVDW : use a neighbour list
every atom keeps a list of nearby atoms within a certain cut off distance (9-12A)
the list is updated every few steps because atoms move during the simulations
only these atoms are considered for interactions which reduces computational cost
Eelectrostatic
need to account for the surrounding medium
permitivity tells you strongly charges are screened compared to vacuum
No NB lis is used: electrostatic interactions don’t decay as rappid
Solution: PME = Particle Mesh Ewald Summation
mesh of charges constructed to treat long range interactions
Solvent representations in MD
Explicit water model
treat every water molecule as an individual entity (with physical properties) within the simulation box
Implicit water model
treat water as a continuous background medium= average effect of water
faster but less accurate
computational power is now big enough to mostly use the explicit water model
Steps in MD: 1-3
add H’s using GROMACS (MD simulation package)
not in X-ray crystal structures because to low electron density
Put the protein into a simulation box = unit cell
computer needs a finite folume to compute interactions and motions
infinite space would be unrealistic and computationally impossible to handle
usually cubic or triclinic
better= rhombic decahedron or truncated octahedron which are smaller than cubic and thus incorporate less water far away from the protein that would need to be computed
PBC = period boundry conditions = copy cell in all directions
this is done to mimmic large solvent environment rather than a tiny droplet
This eliminates edge artifacts, particles at the edge would be in contact with vaccuum leading to surface tension errors
Mainting a constant particle density: for particles leaving at one edge, an identical copy would enter from the other side
Steps in MD: 4-5
Determine the box size
it sould be bigger than the non bonded cut off (9-12A)- but not to large (extra water)
this avoids proteins from interacting or that water molecules in between are attracted by both
Add solvent
Algorithm randomly adds water molecules in a way that agrees with the density of water
Counter ions are added to screen charges and neutralise the system (without charges would accumulate and charges in boxes will repel)
not OH- or H+ because these would alter the pH
Energy minimize to remove steric clashes and relax the solvent
Steps in MD: 6
Start dynamics according to Newtons laws of motion
Essence of MD: if we know the force acting on all atoms we can calculate the a, v and x over time
assign each atom an initial velocity based on th gaussian distribution at a certain T
give us the kinetic energy
Calculate the force on each atom
derivative of the energy, an can be used to calculate acceleration
calculate the speed at the next step
from a and t
calculate the postion at the next step
from vi , ai and t
Energies and coordinates are written to trajectory files
repeating this process generates the MD trajectory
the time step is usually 2 fs = 2 ×10-15 s

leap frog scheme
method that doesn’t try to find position and velocity at the same time but instead ofsets them by half a timestep
calc the velocity at t+1/2Δt
use that to calculate the xi at a full time step ( t+Δt)
use that to jump velocity to the next half step
This creates a balance were errors in first step are often cancelled by the second step - so no accumulation of errors

NVE
The system we’ve now created contains a constant amount of particle (N), cte Volume (V) and constant E = microcanonical ensemble
The total energy is converved
E = Ekin+Epot
particle can move freely constantly exchanging kinetic and potential energy
For our simulation to match conditions of an experiment, we need to swithc our ensemble (most experiments are conducted at constant energy pressure, not energy and volume)
NVE—> NVT: thermostat alters particle velocity so average Ekin corresponds to the target temperature
so system can exhange E with heat bath and E is no longer conserved
NVT —> NVP (barostat) alter the size of the box dynamically so average pressure matches the target value
switching to NVT and NVP ensure the system can reach and remain at thermal equilibrium and reach a physically realistic density
How this fits in an MD workflow
1. System setup (before simulation)
Before any dynamics:
Build the system (structure, box, solvent, ions)
Assign force-field parameters
Choose initial positions and initial velocities
At this point, no ensemble is “running” yet — you just define initial conditions.
2. Equilibration phase
Once the simulation starts:
a) Start in NVE
Initial velocities define the total energy
Particles move according to Newton’s equations
Energy is (ideally) conserved
This step is often very short or implicit, mainly to check stability.
b) Switch to NVT
A thermostat is applied
Particle velocities are adjusted so the average kinetic energy matches the target temperature
The system exchanges energy with a heat bath
Total energy is no longer conserved
Purpose: bring the system to the correct temperature
c) Switch to NPT
A barostat is added
The simulation box size changes dynamically
The system reaches the correct average pressure and density
Purpose: obtain a physically realistic density
3. Production run (after equilibration)
After equilibration:
You typically run long simulations in:
NPT (to mimic experimental conditions),
GROMACS
= MD package that can genereate trajectoreis which record how atom positions, velocities and forces change over time
this can be used to calculate how structural and dynamic poperties evolve over time
this is where analysis begins
e.g. RMSD of a ligand in a binding pocket over time
trajectory vs topology
trajectory describes how thing move
topology contains fixed atom info
How can we use MD
dowload proteins from PDB or use homology model if structure is unknown
carefully investigate input structure: garbe in= garbe out
Also use sufficient heating to explore many conformations on the PES
Run MD simulation
Analysis
RMSD = how protien changes conformation over time from starting point
RMSF = root mean square fluctuation = describe how each atom/ residue in a molecule fluctuates around its average position over time
RG : radius of gyration = how does the molecule change shape
PCA = principle component analysis
Reduces dataset to a small number of principle components with new axes capturing the most important motions an variations in the system
Each PC represents:direction along which atoms move together
eigenvalue: how much motion there is along that direction
applications of MD
conformational changes
analyze physical process of the HIV-1 fusion peptide changes conformation when interacting with the host cell membraneprotein ligand binding
see how interactions evolve, how stable they are, how water and protein flexibility influence bindingprotein folding
only small proteins (µs)
why? correct foldingis essential for function of a protein, misfolding present many diseases
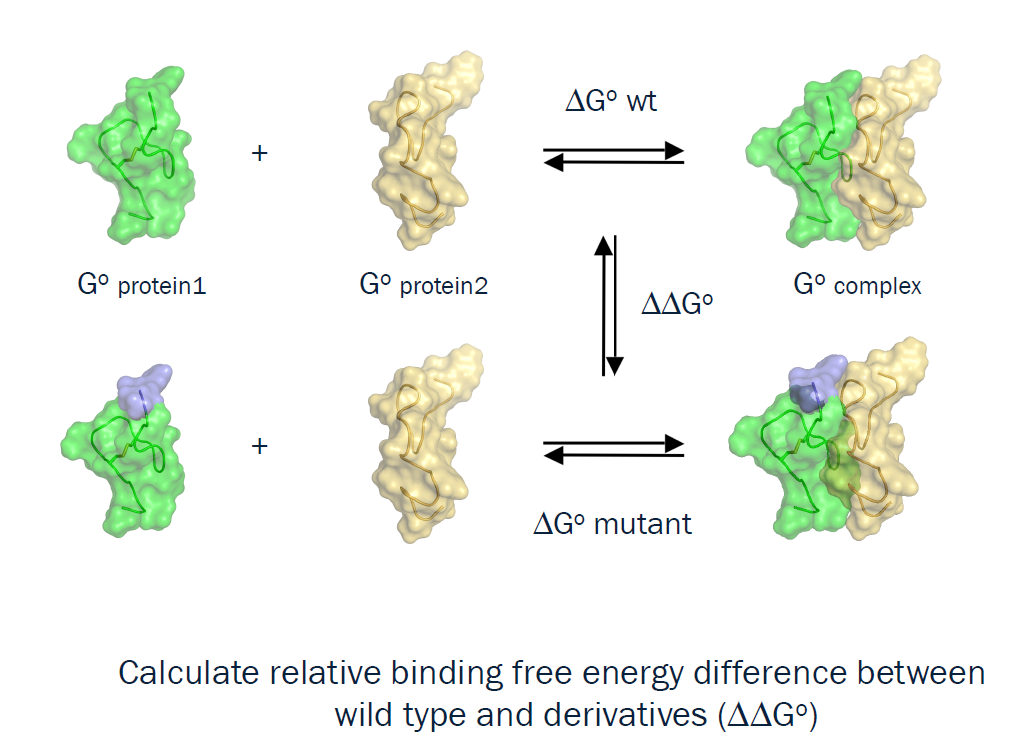
binding free energy calculation