Primary structure protein
1/24
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
25 Terms
What is the limitation of Sanger's method for determining protein sequence? (Big snag)
Sanger's method can only determine the N-terminal connected amino acid (amino acid #1) because hydrolysis destroys the rest of the polypeptide chain.
Who improved Sanger’s method and how?
Edman (Sweden, 1956) improved Sanger's method by developing a technique that allows the N-terminal amino acid to be reacted, removed, and identified without hydrolyzing the peptide bonds. (allowing us to identify amino acid 1,2,3 etc.)
What are the two main steps in Edman Degradation?
Coupling (requires base) – N-terminal amino acid is labeled with PITC.
Cyclization (requires acid) – Cleaves the first peptide bond, releasing the labeled amino acid.
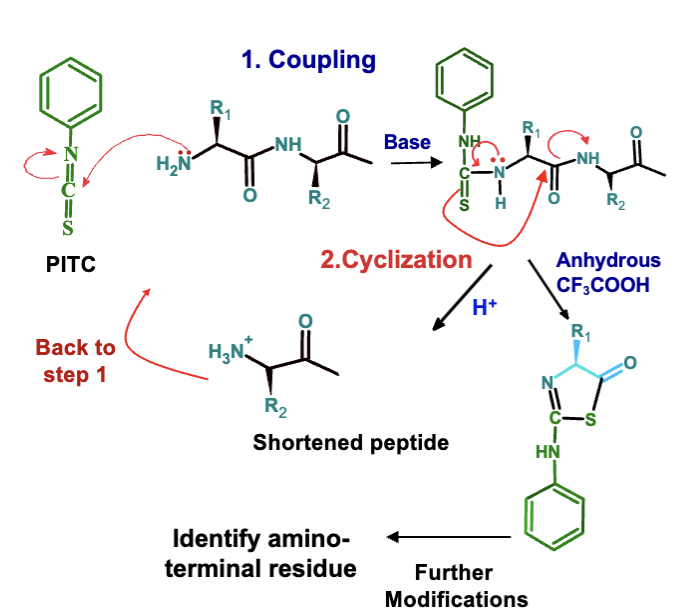
How many cycles can Edman Degradation be repeated, and what is the maximum sequence length it can identify?
The process can be repeated up to 50 times, allowing for the identification of up to a 50 amino acid sequence using automated sequencers.
Why is selective hydrolysis used for studying long protein chains?
It cuts proteins into smaller oligopeptides at specific locations, making sequencing easier (divide and conquer approach).
What enzyme is commonly used for selective hydrolysis of peptides?
Trypsin, a digestive enzyme, which specifically recognizes Arginine (Arg) and Lysine (Lys) side chains. it cuts them at the COO end.
except it can’t be cut if proline follows
Why can’t trypsin cut if Proline follows Arg or Lys?
Proline has a unique ring structure that prevents proper binding in the enzyme’s active site, blocking cleavage.

What’s another enzyme that cleaves peptide bonds
chymotrypsin
Where does chymotrypsin cleave polypeptides
Chymotrypsin cleaves after aromatic residues (Phe (F), Trp (W), Tyr (Y), except when followed by Pro (P).

All fragments end with Phe/Tyr/Trp except C terminal end where we see it ends in Asn
What does cyanogen bromide (CNBr) do to polypeptides?
CNBr cleaves at methionine (Met) residues, breaking the peptide chain on the COO side of Met. It attacks the sulfer atom in Met and converts it to homoserine (Hse).
it can still cut at Met even if Proline follows it
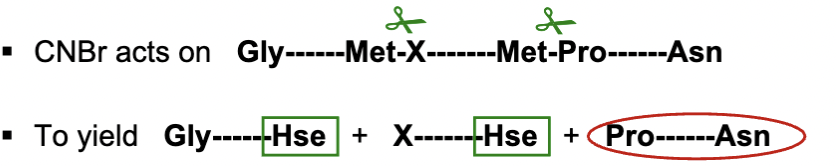
all peptide fragments will have Hse at COO end except for C terminal ebd (where we see Asn)
After digestion with enzymes like trypsin how can we identify the protein?
using mass spectrometry
How is the sequence of an unknown protein determined?
Selective hydrolysis produces oligopeptide fragments.
Chromatography separates these fragments.
Each fragment is sequenced by Edman degradation.
The overlap method is used to reassemble the full sequence.
The overlap method
It reconstructs a full protein sequence by comparing peptide fragments obtained from different cleavage methods (e.g., trypsin & chymotrypsin).
Why is the overlap method necessary?
Because Edman Degradation can only sequence short peptides, the overlap method allows sequencing of longer proteins by aligning smaller fragments.
Given the following peptide sequences obtained after digestion of a protein using trypsin identify the peptide that corresponds to the C-terminus of the original protein.
MET ASN LYS
ASP ILE ALA LYS
GLU ALA LEU PHE ARG
GLU LEU TYR GLN GLY
GLU LEU TYR GLN GLY
because at the C-terminal it wont end in Lys or Arg
Given the following peptide sequences obtained after digestion of a protein using chymotrypsin, identify the peptide that corresponds to the C-terminus of the original protein.
GLN GLY
MET ASN LYS GLU ALA LEU PHE
ARG ASP ILE ALA LYS GLU LEU TYR
GLN GLY
becuase at the C terminus it wont be one with Phe, Tyr, Trp
What is the common method for determining the amino acid sequence of a protein today?
The DNA sequence of the protein gene is determined first using cloning and molecular biology methods, and the protein sequence is predicted using the genetic code.
What is tandem mass spectrometry and what is its advantage?
Tandem mass spectrometry is a technique used to analyze peptides by fragmenting them and measuring the resulting fragments. It only requires tiny amounts of sample, such as a spot from a 2D gel.
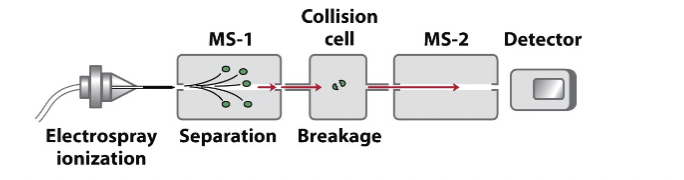
Process of tandem mass spectrometry
Sample Hydrolysis
First MS-1
Collision Cell
Second MS-2
Step 1 Tandem mass spec
Sample Hydrolysis: The sample is hydrolyzed by a protease into peptides. (This occurs before the machine, during sample preparation.)
Step 2 tandem mass spec
First MS-1: Peptides are sorted, and one type is selected to enter the collision cell. (MS-1 stage, where the peptides are analyzed and sorted.)
Step 3 tandem mass spec
Collision Cell: The selected peptides are fragmented at the peptide bond in a random fashion. The breakage of peptide bonds produces b-type (N-terminal fragments) and y-type (C-terminal fragments) peptides.
With trypsin digestion, all y-type fragments will have a charged R (Arg) or K (Lys) at the C-terminus, resulting in a positive charge. (Collision cell, where the peptides are bombarded and fragmented.)
Step 4 tandem mass spec
Second MS-2: The masses of the fragment peptides are measured. Peptides with charges, especially those with R or K at the C-terminus, produce the highest signal in this stage. (MS-2 stage, where the fragmented ions are analyzed for mass.)
Mass spectrometry MS -2 measuring molecular mass
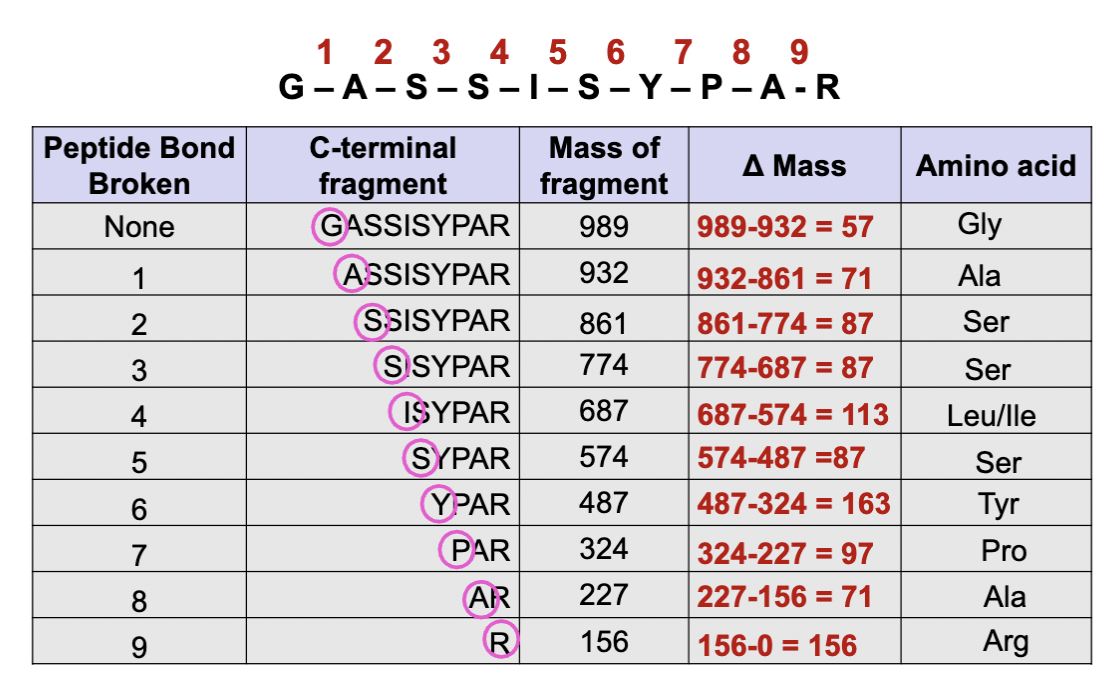
So what we see here is our original peptide is GASSISYPAR
In mass spec, peptide bonds are broken randomly into fragments of varying sizes. The first number 989 represents the mass of the full peptide, and the subsequent numbers represent fragments with one less amino acid.
we calculate the mass differences to identify the amino acid
The smallest one will be Lysine or Arginine because trypsin was used in mass spec, so its at the end of the fragment.
examples of national databases for identifying protein sequences
NCBI
BLAST (compares to all known)