GENOMICS 2.4: Genome sequencing
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
29 Terms
Which is the oldest sequencing method? (first generation)
Sanger sequencing, still used today, based on PCR
How does Sanger sequencing work?
You start with:
dsDNA tempate
Primer that binds to it
DNA pol
dNTPS
A small amount of ddNTP labeled with fluorescence → you run 4 reactions, one for each ddNTP
DNA pol extends the primer, until by chance it adds a ddNTP: it stops the reaction → it will generate many fragments of different lenghts
Separate the fragments by size doing capillary electrophoresis with laser detection → short fragments run faster while long ones slower
Before: gel electrophoresisRead the sequence from smallest to largest

What is a read?
It is the typical maximum nt that can be read in a single experiment → depends on the sequencing technology used
What is the sequence reads lenght of Sanger?
800 bp (between 500 and 900 bp)
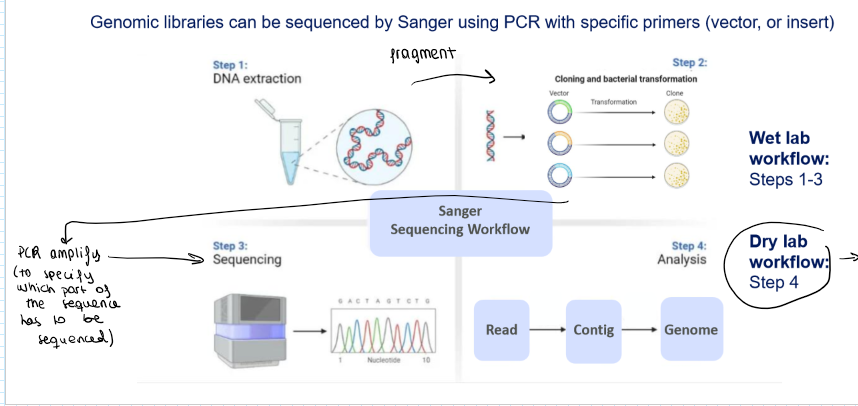
Explain the steps of Sanger Sequencing Workflow for genomic libraries:
Wet lab Workflow:
DNA extraction
Fragment genomic DNA
Clone fragments in vectors and transform in bacteria
PCR amplification
Sanger Sequencing
Dry lab workflow:
Analysis through bioinformatics: read → contig → genome
What instrument do we use to do high-throughput sequencing with the Sanger method?
We use Automated Sequencers → e.g. ABI Prism 3730 DNA Analyzer
The human genome is 3300 Mb → sanger was used to sequence the human genome, it would take a lot of time to do one by one (not possible for the time it took them)
Thanks to automated robots, it made possible the use of microtrays of 96 wells → 96 reacttions at a time, 20 plates for day, 2 Mb per day → to sequence the whole genome it would take 1650 days, if more machines were used in parallel it would take less
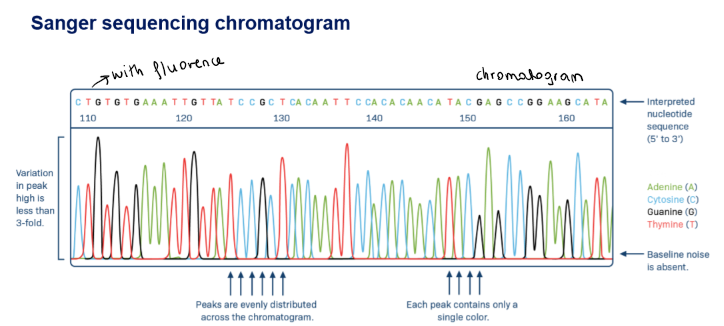
What is the raw data that comes from the sequencing robot?
A chromatogram
What is the process to go from read sequenced into assembled genomes?
Sanger Automated Sequencing
Reading Sequence traces
Contig Assembbly
Genome Assembly
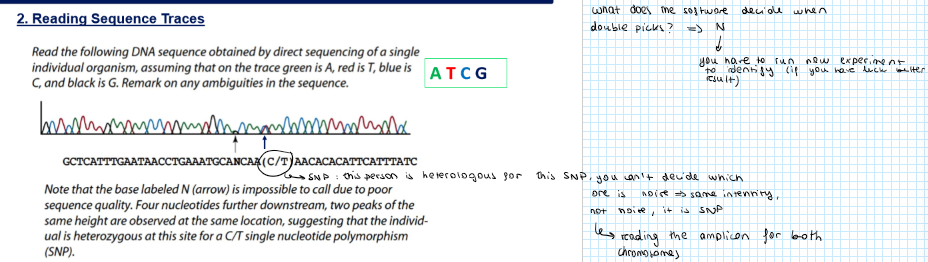
What is automated base calling? (Reading Sequence traces)
It is a software that detects which base represents each peak of the chromatogram → transforms chromatogram to a DNA sequence
The software looks at:
Distance between peaks
Local minima (where one peak ends and another begins)
Peak assignment
Identify double peaks → it can mean lot of things, e.g. SNPs
It defines a quality score for each position (error rate accepted 0,01)
You have to eliminate the first 30 bp (lots of noise) and you have to remove after 800 bp (there is a decline in quality)

What does the software decide when there are double peaks?
The software will respond with N → you have to run a new experiment to identify it
Explain contig assembly:
You go from multiple alignments of sequence reads to assembly of a contiguous DNA sequence (contig)
During this step ambiguities will be resolved and we will reach a sequence consesnus
N spots will be solved by comparing all the data → if you have 3 Gs and 2 Ts, you will decide N is G
Take into account quality scores
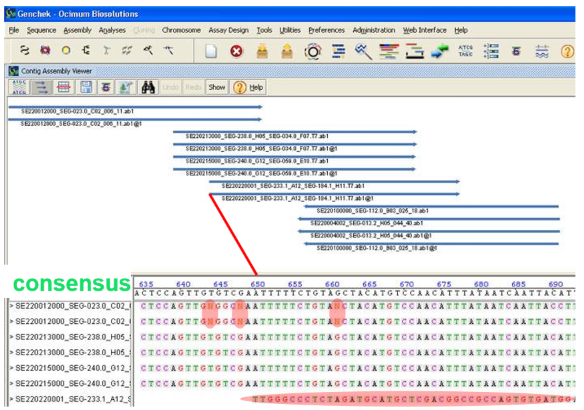
After contig assembly, there will be —
Genome assembly
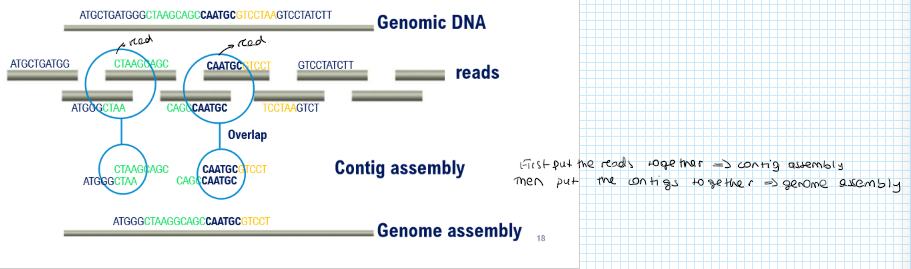
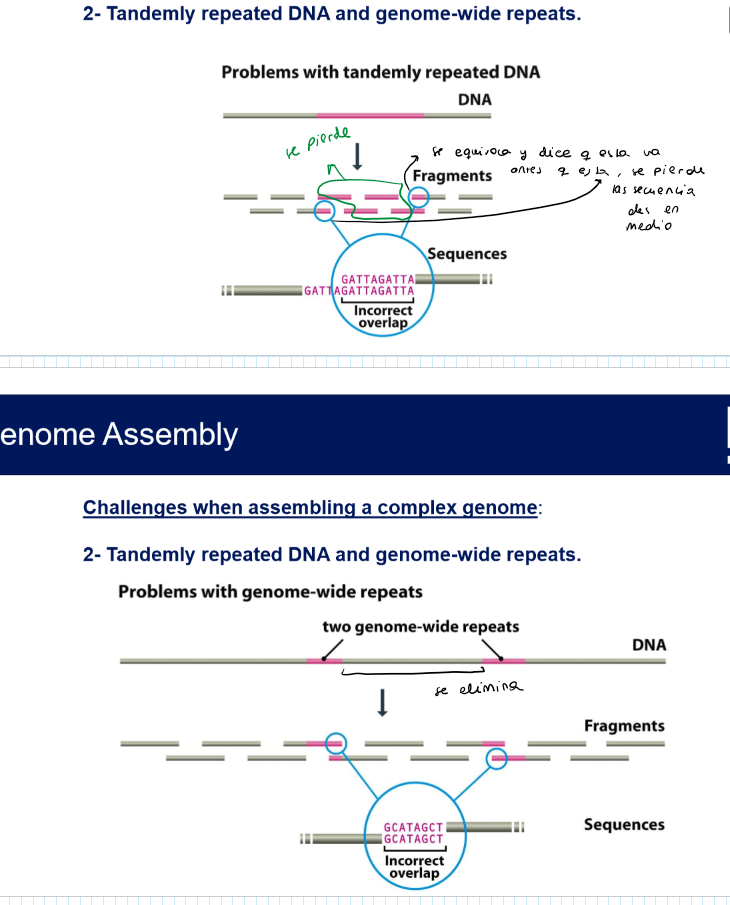
What are the callenges when assembling a complex genome?
Tandemly repeated DNA and genome-wide repeats
What are the different strategies for genome sequencing?
Hierarchical sequencing → International Human Genome Sequencing Consortium
Shotgun sequencing → private iniative: Celera (Craig Venter)
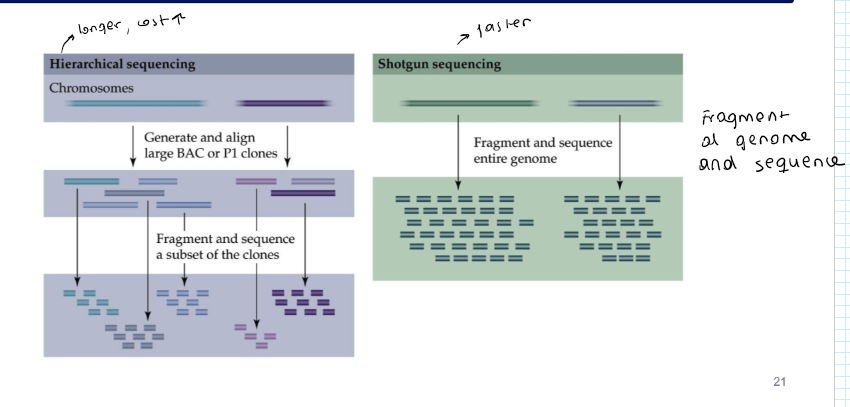
Do hierarchical sequencing and shotgun sequencing use different technologies?
No, they both use Sanger sequencing
What are the steps of Hierarchical Sequencing?
It’s a map based, clone contig, clone by clone strategy → very laborous
Fragment genome in big pieces
Generate BAC library
Physicial or genetic mapping → order the BACs
Markers can be physical or genetic
If the markers are STS (physical marker)
Chromosome walking by:
hybridation: Use the STS as a probe → find BACs that contain it → take a new STS from the end → repeat.
PCR: Use STS‑specific primers → test each BAC → positives overlap → take a new STS from the end → repeat.
Fingerprinting:
You digest each BAC with a restriction enzyme
If two BACs share many fragment sizes → they overlap
If they share none → they don’t
Both can be used together
Fragment BACs in smaller pieces (2 kb) to use Sanger (only the gene of interest, not the whole backbone)
forward and reverse primers (800 bp each → 1,6 kb ideally).
the enzyme restriction sites flanking the gene of interest are the same, so you can use the same primers
Sanger Sequencing → different reads generated (forward and reverse reads)
Assemble reads into contigs
Assemble contigs into the whole genome
What happens if there is a gap between your contigs? You know that contig 1 and 2 are continuous, but there is a sequence in the middle missing (the insert was bigger than 1,6 kb, the primers did not clone all)
You have a sequence gap: you have to design insert primers and try to find the sequence in the library to fill in the gap.
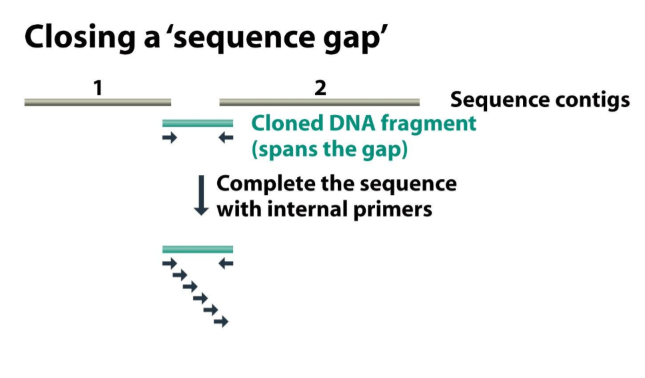
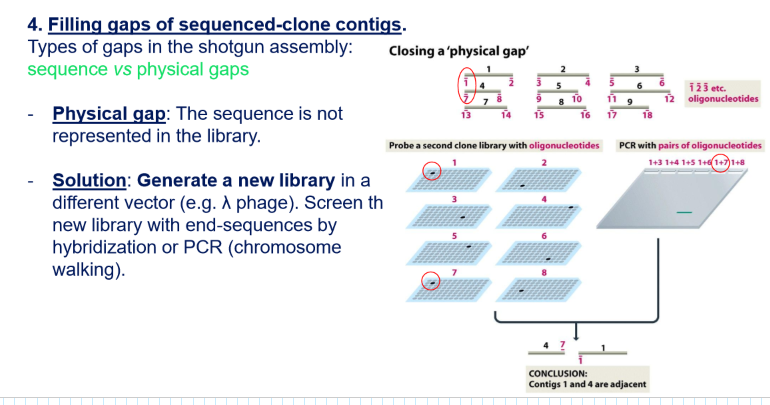
What if the missing sequence is not in your library?
You have a physical gap: you have to start over since the cloning with plasmids → you do a new library with another vector (normally a lambda phage vector) and use again the insert primers to try to find the sequence
What are the advantages of hierarchical sequencing?
Facilitates the correct assembly of tandem repeat sequences
Coverage is uniform across the genome due to sequencing ordered clones
CHAT
"Facilita el ensamblaje correcto de secuencias repetidas en tándem."
Explicación: El genoma está lleno de regiones repetitivas, como si el libro tuviera párrafos que se repiten casi idénticos en diferentes lugares. Si intentas leer el libro entero de una sola vez (como en la secuenciación de genoma completo), cuando encuentres esos párrafos repetidos, no sabrás a qué parte del libro pertenecen.
La ventaja jerárquica: Como ya has ordenado los "capítulos" (clones) antes de leerlos, sabes exactamente en qué parte del libro se encuentra ese párrafo repetido. Es mucho más fácil no perderse.
"La cobertura es uniforme en todo el genoma debido a la secuenciación de clones ordenados."
Explicación: Cuando creas el mapa de "capítulos", te aseguras de que no te falta ninguno y que todos están en orden. Al secuenciar, te comprometes a leer cada "capítulo" por completo.
La ventaja: Esto garantiza que vas a leer absolutamente todo el libro de principio a fin, sin saltarte páginas. No hay zonas del genoma que se lean más que otras; la "cobertura" es pareja.
What are the disadvantages of hierarchical sequencing?
Only minimal clones are sequenced → you don’t sequence the entire BAC library, only the minimal set that covers the genome (low coverage: how many times, on average, each base of the genome is sequenced)
Large initial labor-intensive investment in generating a genomic library and ordering clones to create contig assembly
CHAT
"Cobertura: Solo se secuencia la colección mínima de clones que representan el genoma."
Explicación: Siguiendo la analogía, solo imprimes y lees un único ejemplar de cada "capítulo". Esto es eficiente en teoría, pero...
El problema: Si al leer un capítulo (secuenciar un clon) tienes una duda o hay un error, no tienes otro ejemplar del mismo capítulo para contrastar. En métodos más modernos, tienes muchas copias del libro entero, lo que permite corregir errores por comparación.
"Gran inversión laboriosa inicial en la generación de una biblioteca genómica y el ordenamiento de clones para crear un mapa de clones contiguos."
Explicación: Este es el punto clave. Antes de poder siquiera empezar a "leer" el ADN, tenías que hacer un trabajo titánico y lento.
El problema: Crear la biblioteca de clones (fotocopiar los capítulos) y luego ordenarlos (descifrar el índice del libro) llevaba meses o años de trabajo en el laboratorio, con un coste muy elevado. Era como tener que construir el laboratorio y los instrumentos antes de poder empezar el experimento.
Explain Shotgun Sequencing:
Whole-genome shearing in 2-3 kb fargments
Cloning in a plasmid library
Sequencing with 5-10 fold redundancy (coverage: how many times, on average, each base of the genome is sequenced.)
Assembly requires high computational capacity
Why does whole-genome shotgun sequencing need high computational sequencing?
2 Mb/day: human genome in about 16500 days → 165 day if 100 sequencers
That is why Celera used lots of machines running in parallel

Why was the assembly a two-step process in whole-genome shotgun sequencing?
The shotgun strategy was not very good for repetive DNA (they did not have physical maps)
Initial phase: successful assembly of up to 90% of the genome, repetitive sequences were left out
Finishing phase: very laborious, it requires new genomic libraries to solve:
Assembly of repetitive sequences
Closing sequence and physical gaps → more dificult because they are bigger gaps
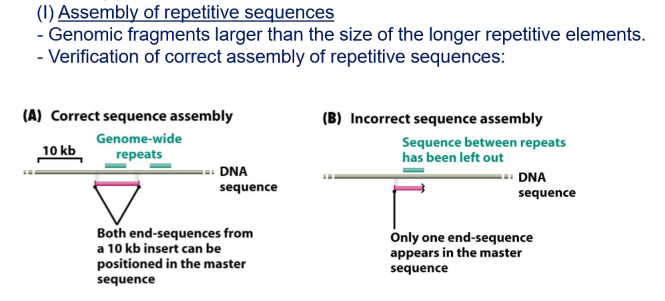
How did they verify the assembly of repetitive sequences in the assembly of shotgun sequencing?
They used public information from the other project to design forward and reverse primers of the repeats, if they did not find the product in the final assembly it meant that they had lost that sequence
What is a scaffold?
In genomic mapping, a series of contigs that are in the right order but not becessarily connected in one continous stretxh of sequence
How did they solve gaps in the assembly of shotgun sequencing?
Sequence gaps closed by further sequencing of individual clones
A region is present in the plasmid library
But you didn’t sequence enough clones
Or the reads didn’t overlap enough
Or the region is hard to sequence (GC‑rich, repeats, etc.)
Physical gaps are difficult to close: require analysis of 100 kb fragment library → they needed to generate BAC libraries to try and close the gaps
A region of the genome is missing entirely from the plasmid library
BACs (100–300 kb inserts) are much more stable and represent the genome more completely.

What are the advantages of whole-genome shotgun sequencing?
Much faster in assembling a draft with 90% of the genome
What are the limitations of whole-genome shotgun sequencing?
Requires high computational capacity
Completeness of the genome: the fisnishing phase is very labor intensive → BAC libraries must be generated to close physical gaps
Accuracy: coverage is not uniform across the genome due to the random nature of the process
Comparison between Hierarchical shotgun sequencing and Whole genome shotgun sequencing: