PHARM 203 - MIDTERM
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
88 Terms
Pharmaco——- is the study of what the drug do to your body.
Pharmaco——- is the study of what your body does to the drug.
dynamic, kinetic
Most drugs bind to a ——- protein (i.e. receptors, ion channels, enzymes, transporters).
regulatory
Binding is the term used to describe the physical act of a small molecule joining with a protein target to form a ——.
complex
Drugs often bind to a protein target at the same site as the ——— ——-.
endogenous ligand
A ligand which binds to a receptor protein and activates the receptor is called an ——.
A ligand which binds to a receptor and does NOT activate the receptor, but prevents an agonist from binding and activating the receptor, is called an ——-.
agonist, antagonist
Almost all endogenous ligands are ———.
The majority of drugs that bind to receptors are ——.
agonists, antagonists
The property of an agonist that allows it to “turn on” a receptor is called the ——- of the agonist.
efficacy
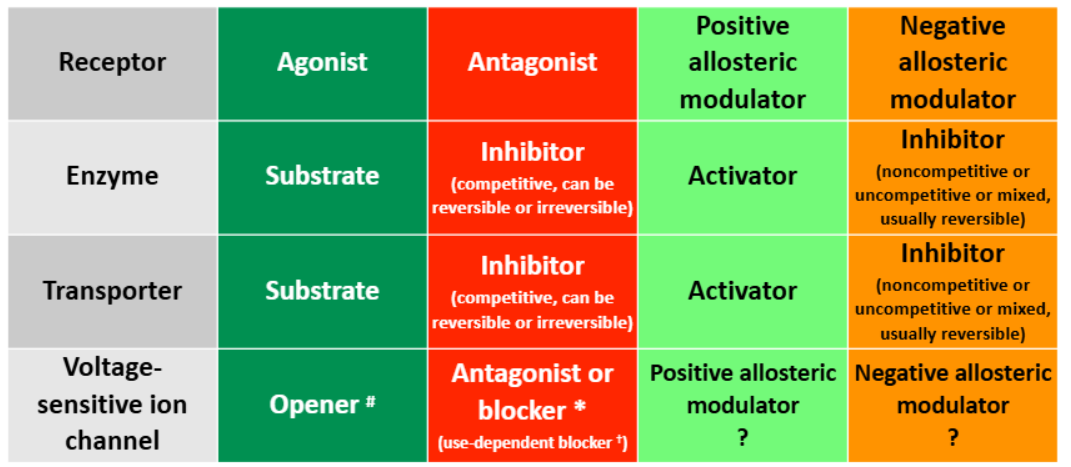
There —— (is/is not) an agonist binding site on voltage-sensitive ion channels.
is not
——- ——- bind to a different site from the agonist binding site, and modify either agonist binding or the receptor’s response to an agonist.
allosteric modulators
Bay K8644 is an experimental L-type Ca2+ channel ——- which binds to the nifedipine binding site, evokes Ca2+ flux and has a positive inotropic effect. It forces the channel to open even when there’s no voltage change.
Nifedipine is an L-type Ca2+ channel ——- used to treat hypertension and angina. It prevents Ca2+ influx even in change of voltage.
opener, antagonist
Lidocaine is a —— ——- ——- of voltage-activated Na+ channel that physically blocks the channel. It is used as a general anesthetic.
use-dependent blocker
——- refers to the force of muscular contraction (particularly in cardiac muscle). ——- ——- increases the force of contract, ——- ——- decreases force of contraction.
inotropic, positive inotrope, negative inotrope
Drugs bind to proteins through ——-based and —— interactions.
charge, hydrophobic
The higher the ——- and —— of complimentary interactions, the more tightly the drug binds.
number, quality
What factors can reduce binding tightness or can prevent binding altogether?
shape (steric hindrance)
lipophilic/hydrophilic incompatibility
charge clashes
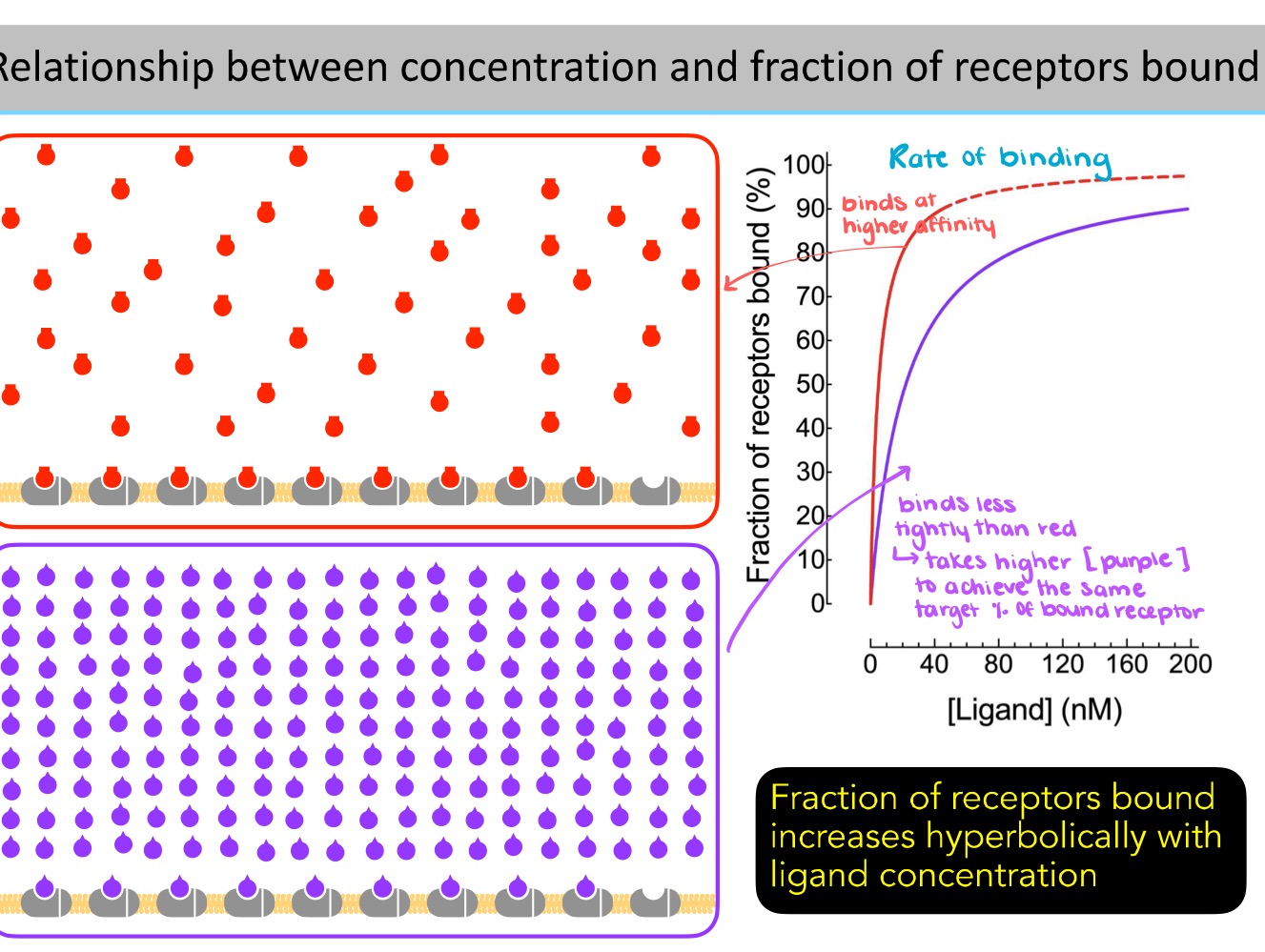
Fraction of the receptors bound increases ——- with ligand concentration.
hyperbolically
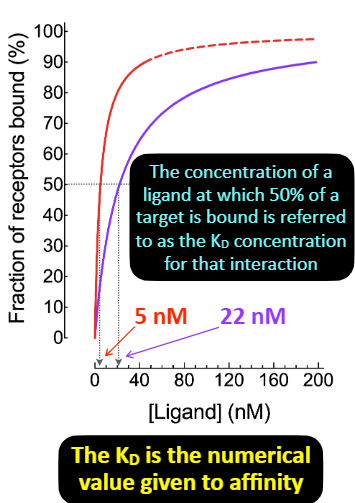
——— describes the tightness of binding.
The affinity of a ligand to a particular protein can be quantified using a value called ——. It represents the concentration of a ligand at which 50% of target is bound.
affinity, KD
The drug concentration that results in 50% of targets present being bound at equilibrium is referred to as the drug’s ——- concentration for that target.
KD

The equation for a hyperbola that relates ligand concentration to binding is called the ——- ——- equation.
Hill-Langmuir
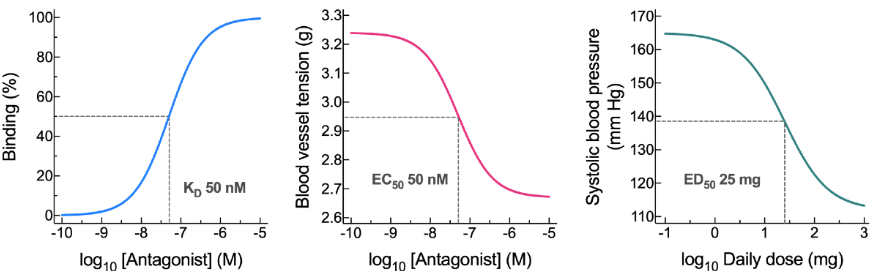
A ligand binding study generates a —— curve. This gives us the —— value → concentration of ligand at which 50% of targets are bound.
An isolated tissue study generates a ——- ——— curve. This gives us the ——- value → concentration of ligand that gives 50% of the maximum effect.
An in vivo study generates —— ——- curve. This gives us the —— value → dose of ligand that generates 50% of the maximum response.
These are all —— curves achieved by taking the log of the hyperbolic curve.
binding, KD, concentration-effect, EC50, dose-response ED50, sigmoidal
The data for a —— dose response curves can be obtained from a single patient at different doses.
While the data for a —— dose response curve are from a group of patients - where the effect either does, or does not occur (i.e. anesthetics - dose that makes the patient awake vs. not awake).
graded, quantal
The ——- rate (kon) measures how efficiently and rapidly ligand binds to the receptor.
The —— rate (koff) measures how long the ligand stays bound before leaving the receptor.
These rates affects the —— of the ligand to the receptor.
association, dissociation, affinity
A ligand with a larger dissociation rate constant for a target is —— likely to associate with the target when in the vicinity of the target.
more
A ligand with a larger dissociation rate constant for a target will remain bound to the target for —— time before dissociating.
less
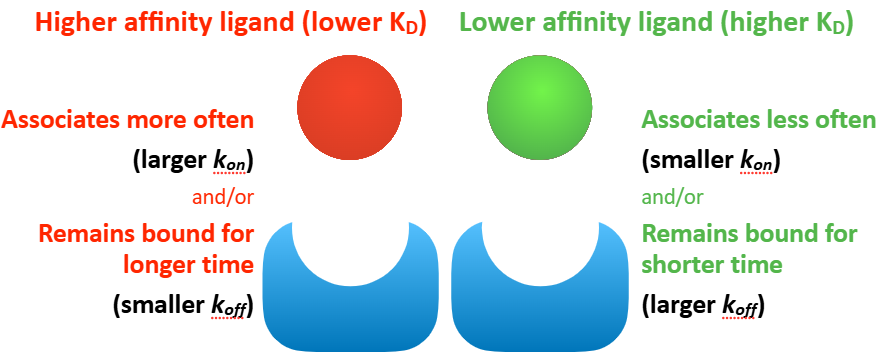
Describe the KD, kon, and koff and what they mean for a higher affinity ligand vs. lower affinity ligand.
higher affinity:
lower KD - require lower [ligand] to reach 50% target occupancy
larger kon - associates more often
smaller koff - remains bound for longer
lower affinity:
higher KD - require higher [ligand] to reach 50% target occupancy
smaller kon - associates less often
larger koff - remains bound for shorter time
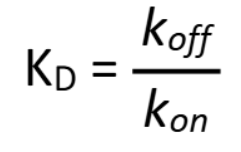
How do you calculate KD from kon and koff?
KD = koff/kon
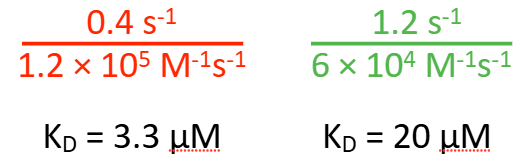
Which of the following do you expect to have a higher affinity for the specified ligand?
red
At —— ——, the association rate = dissociation rate.
binding equilibrium
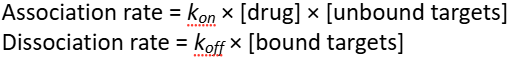
Increasing the drug concentration directly ——— the drug association rate and indirectly —— the drug dissociation rate.
increases, decreases
Drugs with very large kon values are referred to as ——- ——- drugs. These drugs are more likely to bind to a target in the vicinity after dissociating than to diffuse away, allowing lower doses to be used and reducing the chances of side-effects.
fast-on
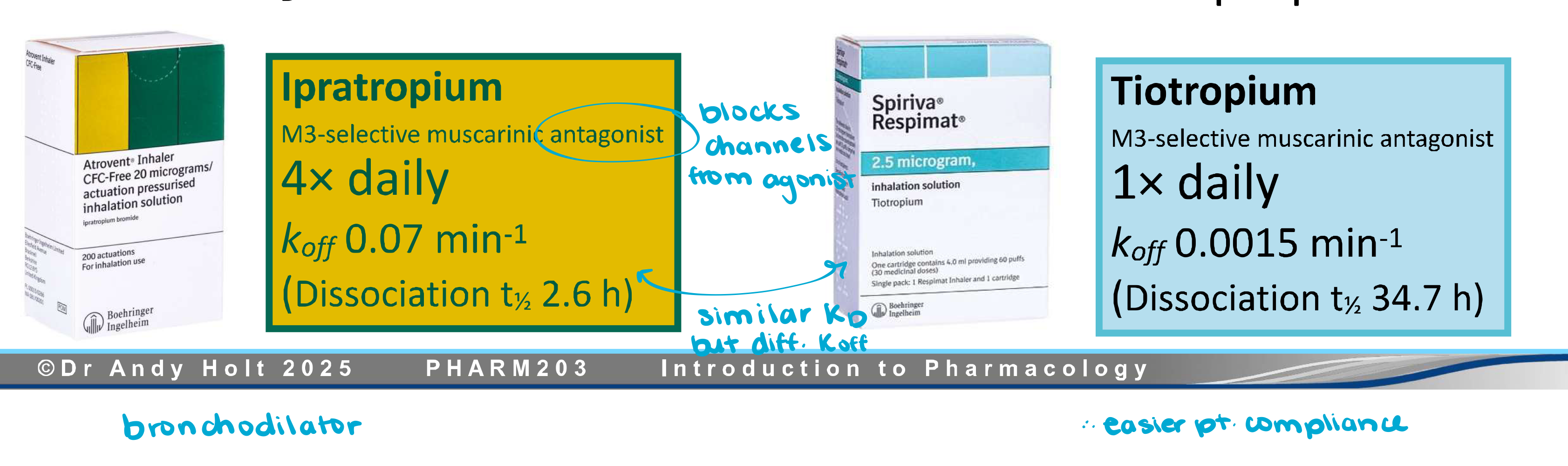
Drugs with very small koff values are referred to as —- —- dugs. The drug effects can persist long after most of the drug has been cleared from the body, allowing less-frequent dosing.
slow-off
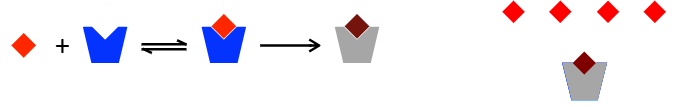
Most irreversible drugs are —- ——. Where the initial interaction is reversible, followed by covalent bonding.
The protein is permanently inactive, and requires new enzymes to be made to recover activity.
enzyme inhibitors
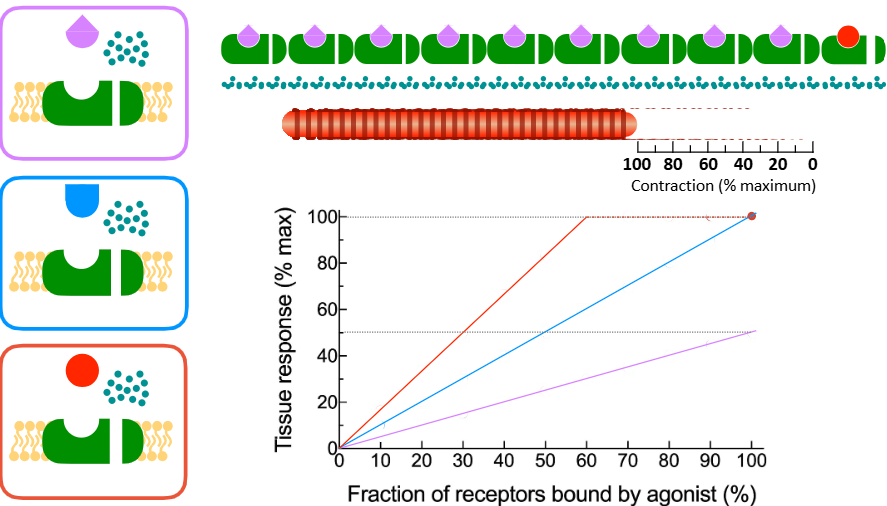
Agonist —— refers to the effectiveness of an agonist in activating a single receptor.
i.e. higher efficacy agonist - cause stronger response
efficacy
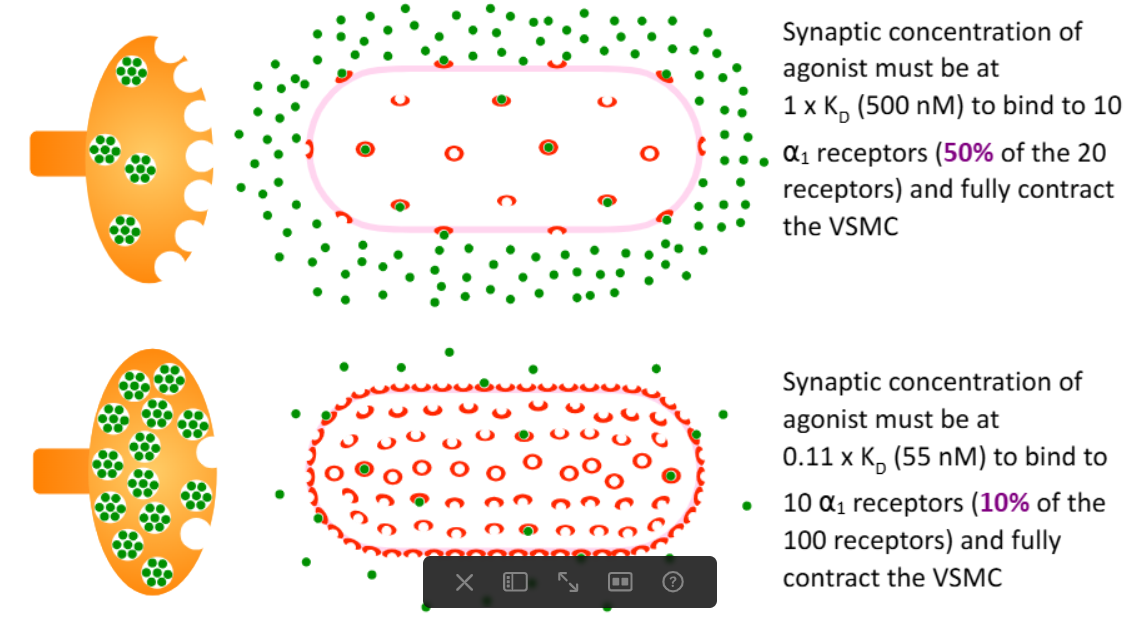
How is agonist efficacy related to the number of receptors on the cell/tissue?
the same agonist acting at the same type of receptors will have higher efficacy in a tissue that expresses a higher # of receptors
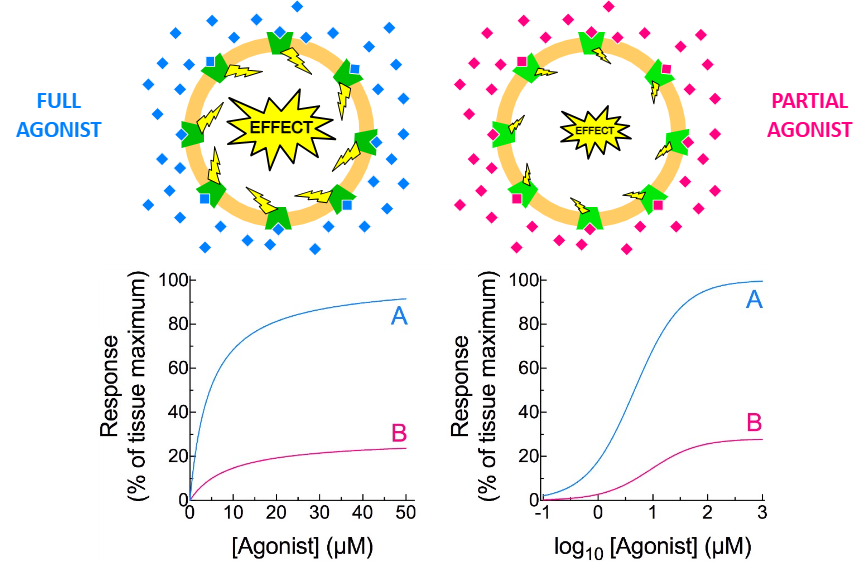
A —— agonist is one that causes a less than maximum possible effect.
partial
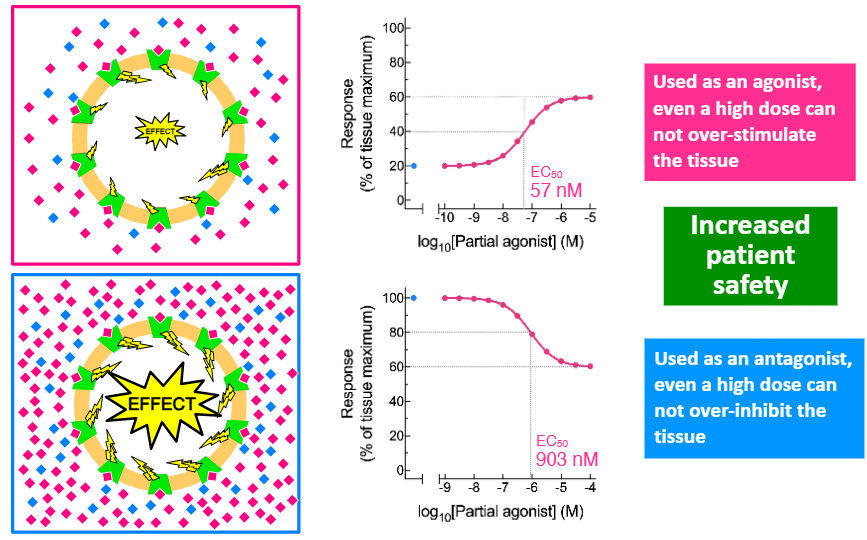
Clinically, partial agonists are used as agonists AND antagonists.
This can help to increase —— —-.
patient safety
Drug —— is the dose of a drug required to achieve a particular magnitude of response.
They are often compared using —— values.
potency, ED50
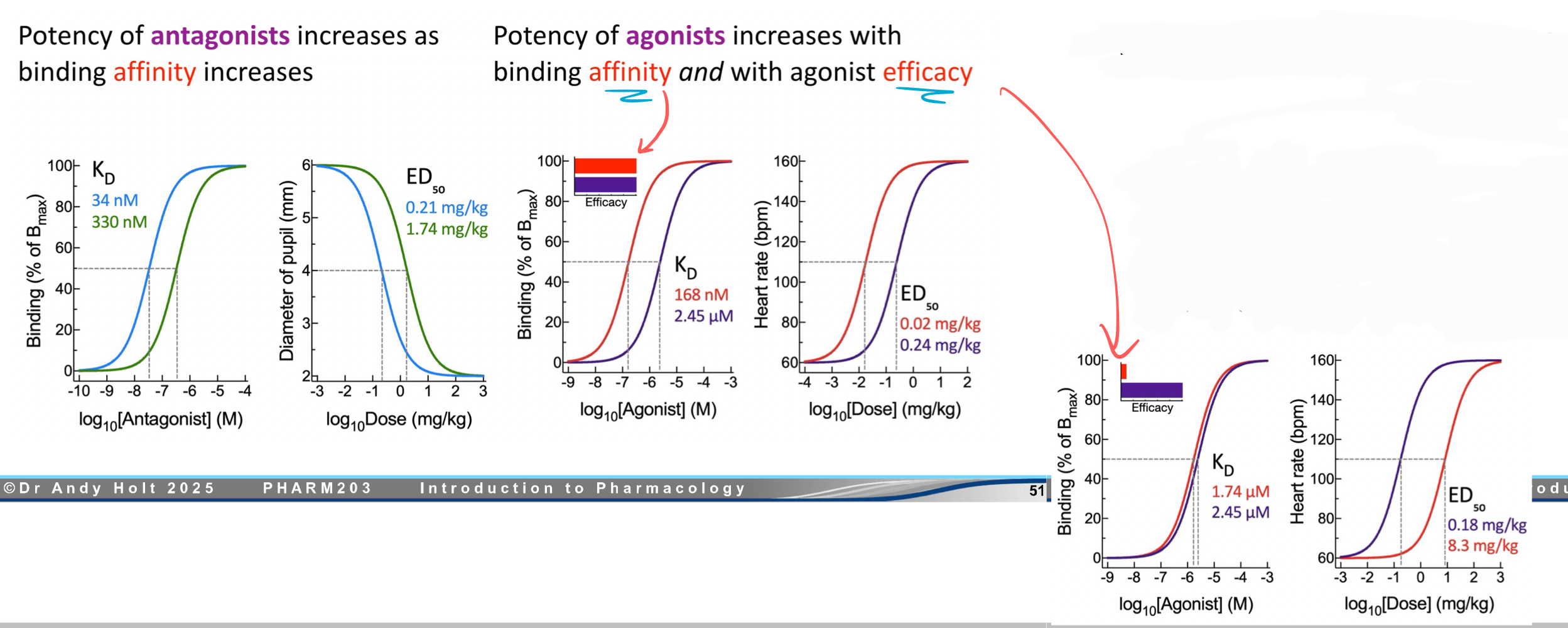
The potency of —— increases as binding affinity increases.
The potency of —— increases with binding affinity AND with agonist efficacy.
antagonist, agonist
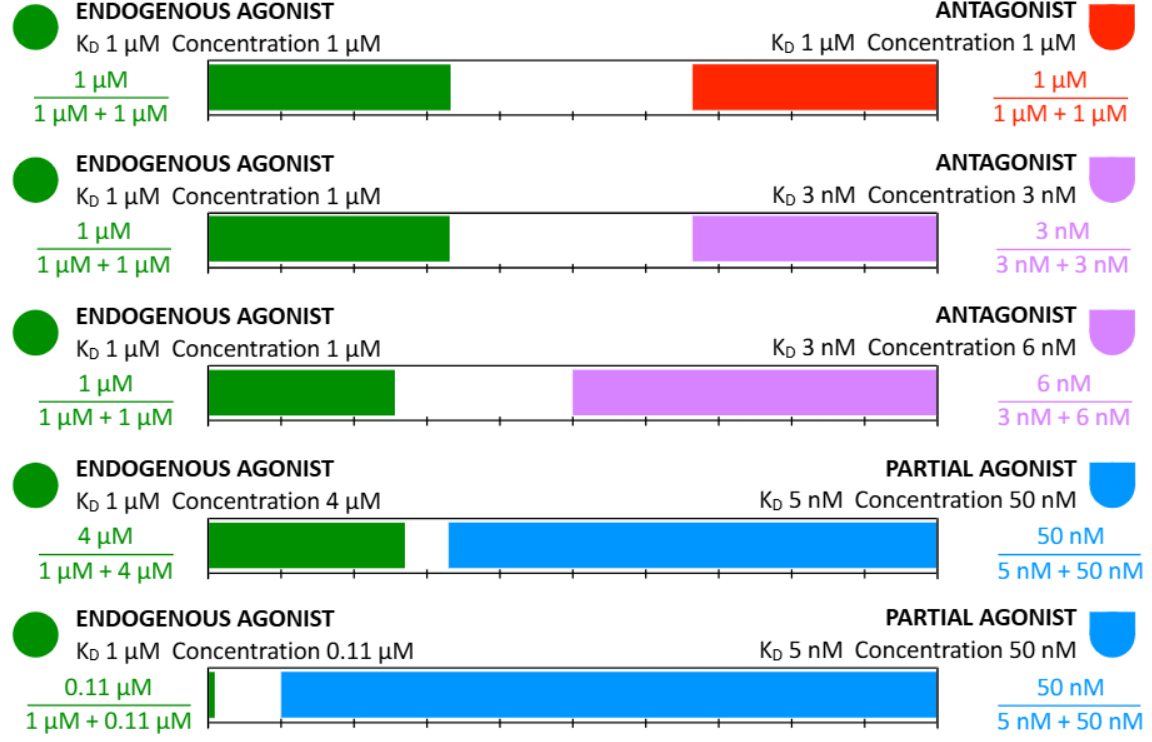
Competition occurs when two or more ligands are present that bind to the same protein, with the degree of binding of each to the available targets determined by the ligand concentration relative to its ——- for that protein.
KD
The “main” binding site is called the —— binding site. Whereas an alternative alternative site is called —— binding site.
orthosteric, allosteric
What does ADME stand for?
absorption, distribution, metabolism, excretion
ADME determines the drug’s plasma ——— versus —— ——.
i.e. how quickly the drug reach an effective concentration at the target site
how long the drug remain at an effective concentration
concentration, time profile
——— is the net movement of a drug from the site of administration into the bloodstream.
——— is the net movement of drug from the bloodstream into the organs and tissues.
absorption, distribution
——— drugs are administered by mouth (PO) or rectal (PR). These drugs must pass through the —— before it enters systemic circulation and there is often drug loss from this.
enteral, liver
——— drugs can be administered through intravenous injection (IV), intramuscular injection (IM), subcutaneous injection (SC), topically (transdermal, inhalation, insufflation, eye/eardrops), buccal (BUC), or sublingual (SL).
These drugs bypass the liver and are absorbed straight into the bloodstream.
parenteral
Drugs administered through the rectum (PR) can also be absorbed ——- through the blood vessels of the rectum (bypass liver).
parenterally
What are some formulation techniques used to optimize absorption?
drug particle size
protective coatings
slow and fast release pellets, polymers, osmotic technology
depot injection
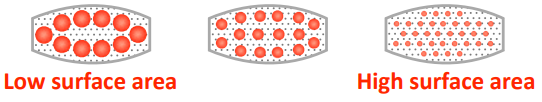
The smaller the drug particle size, the ——- the surface area, the —— the drug dissolves.
higher, faster
——— ——— (SR) prolongs the time for which the drug leaves its carrier.
——— ——— (CR) releases the drug at a constant rate.
Both SR and CR are considered ——— ——— (ER, XR).
sustained release, controlled release, extended release
Describe the flow of a drug that is absorbed through the stomach/intestines before it reaches systemic circulation.
drain from walls of stomach/intestines → portal vein → liver → systemic circulation
The loss of drug to metabolism on its first passage through the liver is called —— —— ——-.
first pass metabolism
The fraction of an oral dose of drug that reaches systemic circulation as “intact drug” is referred to as —— ——.
oral bioavailability
A dose of 20 mg was taken orally. 16 mg of the drug remains after the gut and 4 mg remains after passing through the liver. What is the:
Fraction escaping gut (fg)
Fraction escaping liver (fH)
Hepatic extraction ratio (EH)
Oral availability (F)
0.8
0.25
0.75
0.2
Charged drugs can diffuse across the lipid bilayer. T/F
F
Most acids are —— acids or bases where they can exist in a charged form. It must be ——- to cross the bilayer, and layer protonated/deprotonated.
weak, uncharged
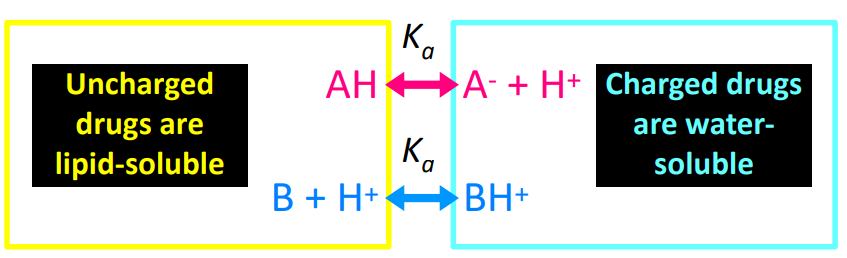
——- is the pH at which 50% of the drug is ionized.
pKa
Weak ——- are rapidly and extensively absorbed from the stomach.
Weak ——- are better absorbed from the small intestine.
acid, base
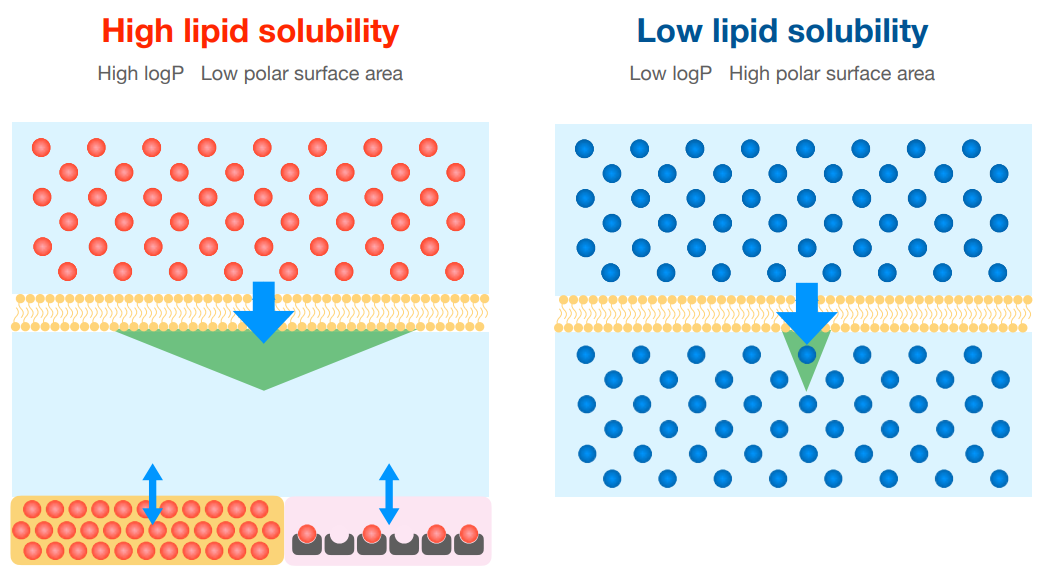
Most drugs are far more ——- than ——- soluble.
lipid, water
A ——- logP or ——- polar surface area (PSA) indicates that the drug is more lipid soluble.
higher, lower
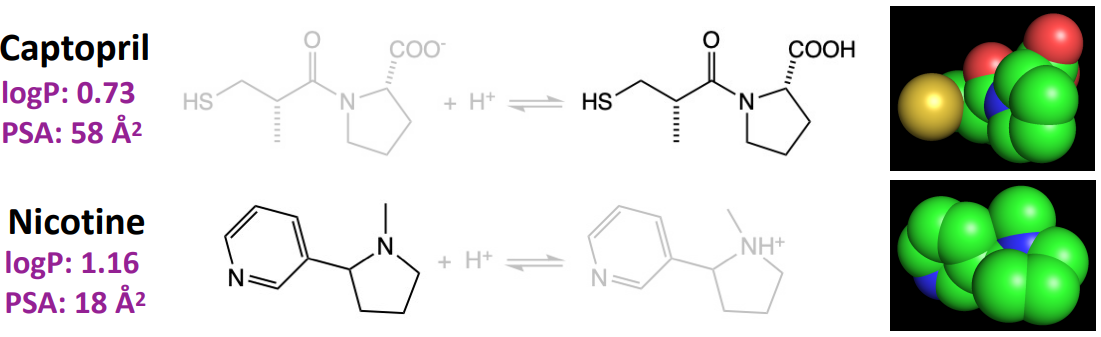
Which drug is more lipid-soluble? Captopril or Nicotine?
nicotine
“A drug will flow from an area of higher concentration to an area of
lower concentration, with the rate of flow being higher with larger
concentration gradients” is known as —— —— of ——-.
Fick’s Law of Diffusion
Which of the following would take a shorter amount of time to cross the lipid membrane?
a) high lipid solubility
b) low lipid solubility
a
What other factor can impact how rapidly drugs distribute into the tissue and reach equilibrium?
level of vascularization
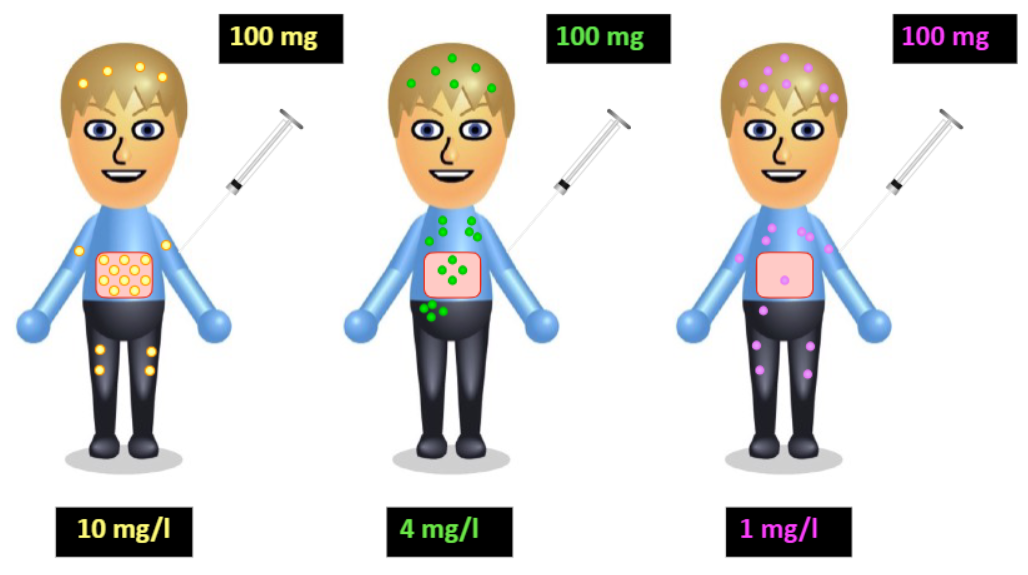
The more extensively a drug distributes from the plasma into the tissues, the ——- the amount of the administered dose remains in the plasma, and therefore the —— the concentration of drug remaining in the plasma.
smaller, lower
The —— —— of ——- (AVD or VD) is the apparent volume of plasma that would contain the total body content of the drug at a concentration equal to that in the plasma.
Basically, it tell you how much of the drug has been distributed into the tissues. A higher AVD means a higher level of drug distribution.
apparent volume of distribution
If given logP = 1.5 and 30:1 ratio. What does the ratio tell you about the drug?
30:1 is the oil to water solubility
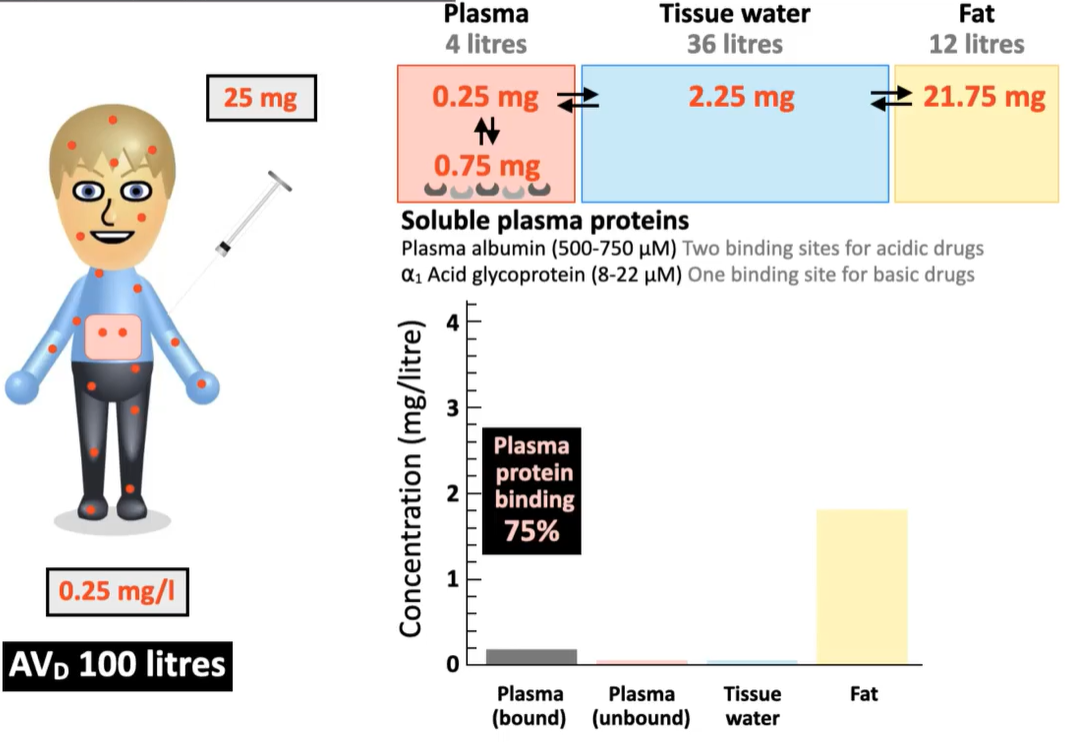
A 25 mg dose of a drug is administered to a young, health patient (4 L plasma). After drug distribution, 0.25 mg of the drug remains unbound in the plasma, and 0.75 mg are bound by plasma protein. The rest has been distributed into tissue water and fat in the body.
What is the drug concentration in the plasma? What is the AVD?
[drug in plasma] = (0.25 mg + 0.75 mg) / 4 L = 0.25 mg/L
AVD = 25 mg x 1L/0.25 mg = 100 L

Based on the following information, what is the concentration of unbound drugs in the tissues that can bind to target receptors?
2.25 mg/L x 36 L = 0.0625 mg/L
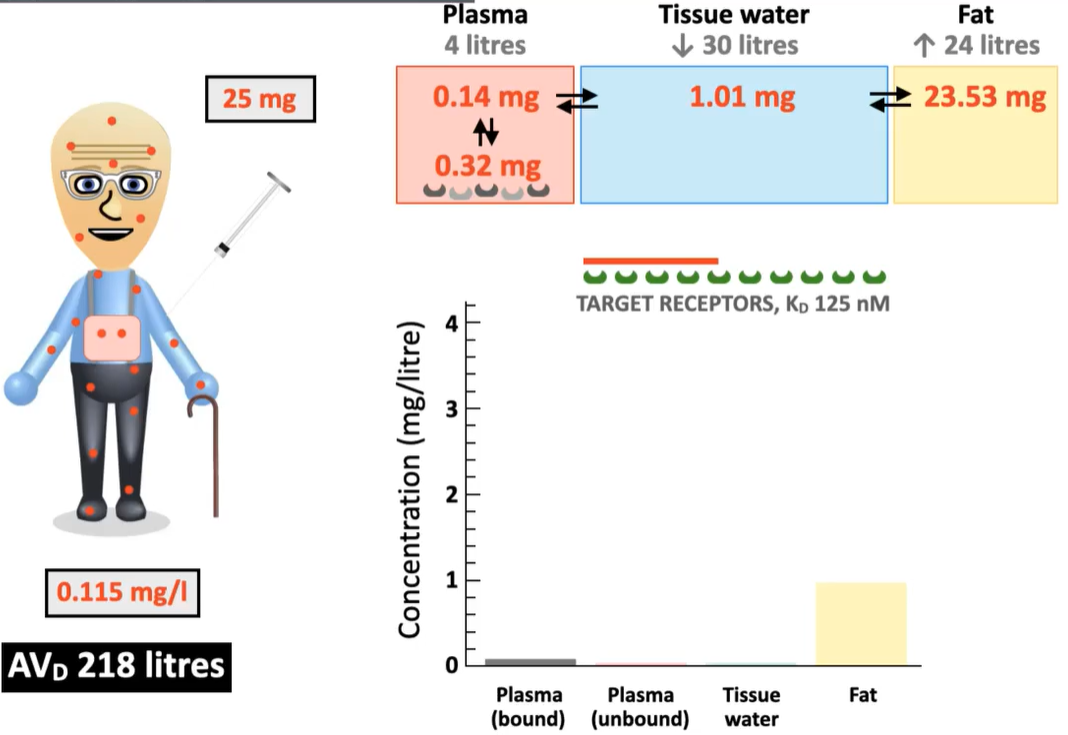
If the same drug is administered into an elderly patient. What are the changes in terms of:
portion of drug dissolved in fat content vs. remaining in plasma?
% of drug that can bind to target receptors?
Should a higher dose be administered to the elderly patient?
higher portion of drug is dissolved in fat content
less remaining in plasma and can bind to target proteins
no - do not give elderly pt a higher dose because drug will likely take longer to be eliminated from the body
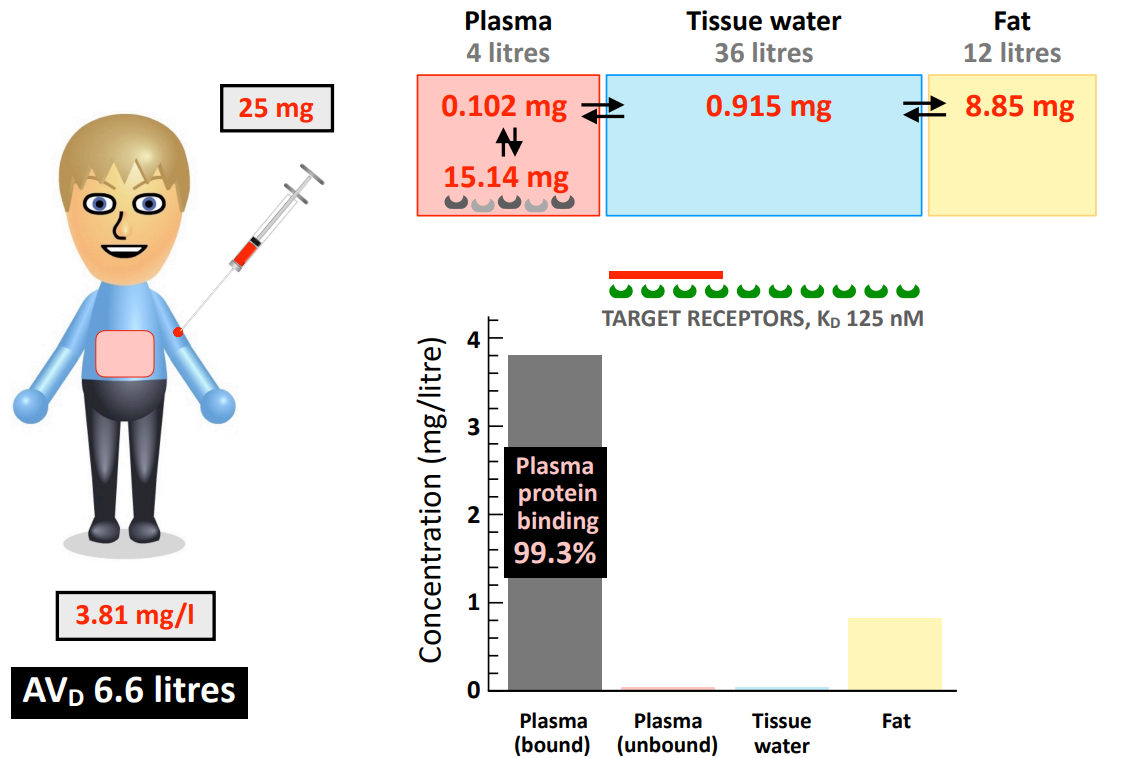
What does a low AVD value (close to 4 L plasma volume) tell you about the drug distribution?
most of the drug is retained in the plasma due (extensively bound to plasma protein) → lower distribution to body tissues
—— is the irreversible removal of a drug compound from the blood so that it is no longer able to exert a pharmacological effect.
elimination
Elimination is divided into 2 categories:
—— is the elimination of unchanged drug into urine, feces, expired air, sweat, etc.
—— —— is enzymatic metabolism of the parent drug into a different chemical compound (metabolite).
excretion, enzymatic metabolism
Metabolism predominantly occurs via —— —— (in the ——).
Excretion predominantly occurs via —— —— (in the ——).
hepatic metabolism, liver
renal elimination, kidneys
—— —— (CI) refers to the volume of plasma cleared of drug in unit time (mL/min or L/hr) due to elimination.
drug clearance
How do you calculate total body clearance?
total body clearance = renal clearance + hepatic clearance
—— —— —— (E) is the proportion of drug removed by a single transit of that drug through the organ (0 ≤ E ≤ 1).
organ extraction ratio
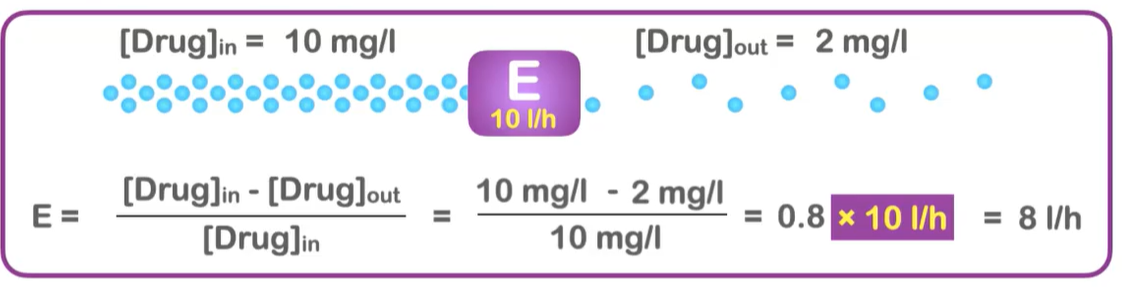
![<p>If [drug]<sub>in</sub> = 10 mg/L and [drug]<sub>out</sub> = 2 mg/L. What is the organ extraction ratio (E)?</p>](https://knowt-user-attachments.s3.amazonaws.com/18698246-78cd-4f52-ab89-76add0c3e339.png)
If [drug]in = 10 mg/L and [drug]out = 2 mg/L. What is the organ extraction ratio (E)?
0.8
What is the equation for clearance by an organ?
CI (drug clearance) = Q (blood flow through organ) x E (organ extraction ratio)
What is the hepatic clearance (QH) in a healthy adult?
90 L/hr
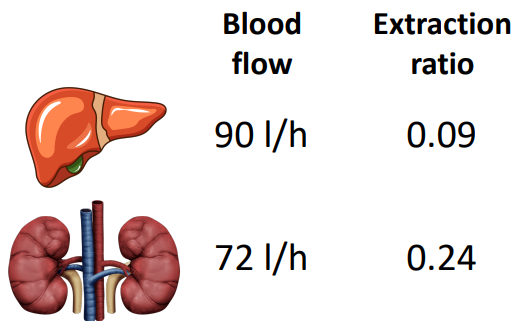
Based on the following information, what is CIH, CIR, and total body clearance?
CIH = 90 L/hr x 0.09 = 8 L/hr
CIR = 72 L/hr x 0.24 = 17 L/hr
total body clearance = 8 L/hr + 17 L/hr = 25 L/hr
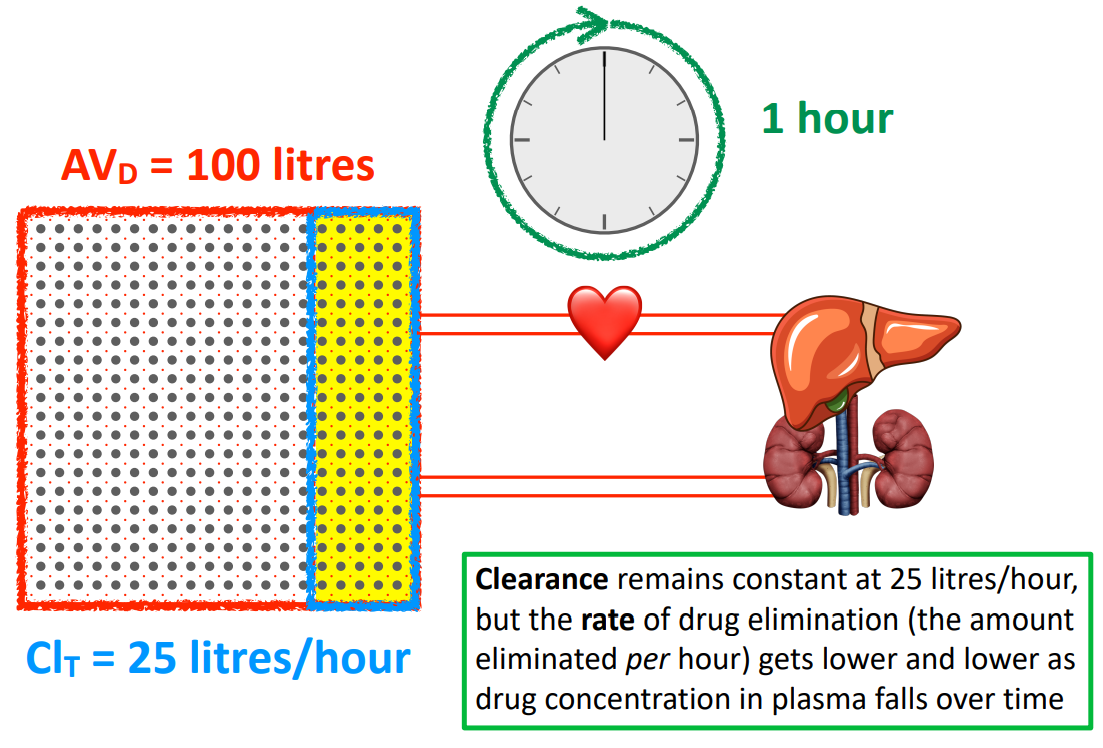
If 100 L of plasma are cleared at a rate of 25 L/hr for a drug. It will take 4 hours to completely clear the drug. T/F
F
clearance remains constant at 25 L/hr, but as the drug leaves, the drug molecules remaining redistributes and its concentration in the plasma decreases
therefore, the rate of drug elimination (amount eliminated per hour) also decreases as the drug concentration in the plasma decrease over time (will take longer than 4 hours)
Only drugs that are —— by plasma proteins can be filtered and secreted.
unbound
——soluble drugs can be reabsorbed back into the bloodstream (tubular reabsorption), while —— drugs remain in the urine and become concentrated.
lipid, charged
What are 3 factors that affect renal clearance?
GFR - glomerular filtration rate (changes with age, sex, disease)
fu - fraction of unbound drug (number of plasma protein change with age, disease)
FReabs - fraction of drug reabsorbed (change with urinary pH)