Fisiologia do Sangue
1/28
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
29 Terms
Sangue
Único tecido fluido do nosso organismo;
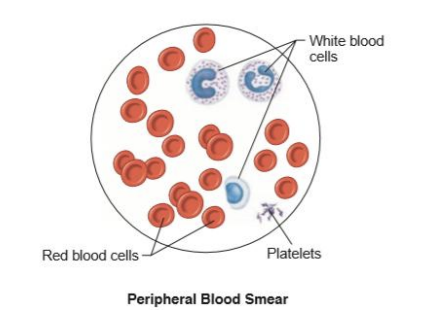
É constituido por hemácias, leucócitos e plaquetas → suspensas
É um fluido não-Newtoniano, cuja reologia é muito influêncida:
Temperatura do corpo
Colesterol total: lipoproteinas aumenta, maior a viscosidade do sangue
Hematócrito: percentagem de volume ocupado pelos glóbulos vermelhos no volume total do sangue
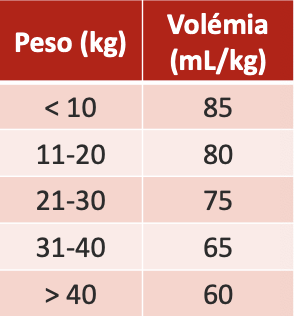
Volémia= volume total do sangue circulante
Funções do sangue
Transporte: gases, hormonas, calor, metabolitos e nutrientes
Proteção: criada pelos leucócitos, contra hemorragia (hemostase)
Diversos papéis na inflamação
Neutralização e destruição de agentes patogénicos (ex: microrganismos e células tumorais)
Hemostase
Regulação: Equilíbrio hidroeletrolítico e ácido-base
Orgãos metabolicamente mais ativos: os músculos
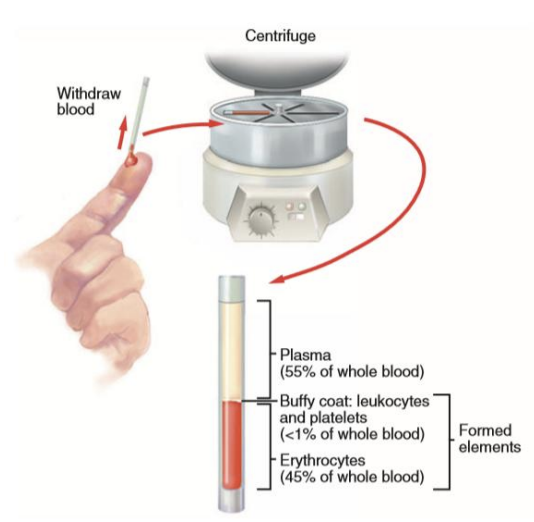
Constituição do sangue
fase sólida = elementos figurados (45%)
≈45% hemácias <1% leucócitos
fase liquida = plasma (55%)
10% elementos sólidos
90% água
Hemácias
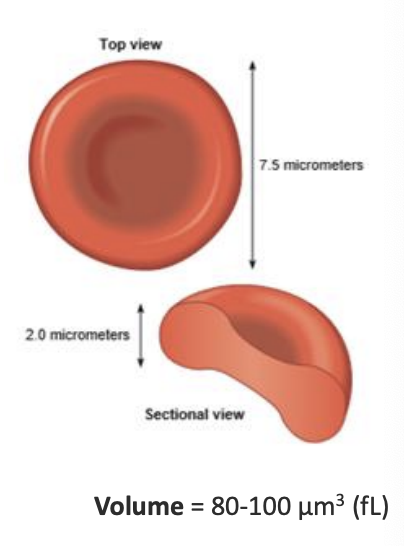
Hemácias = Eritrócitos = Glóbulos vermelhos
forma de discos bicôncavos
sem núcleo, mitocôndrias ou ribossomas
vivem 120 dias
transporte de oxigênio → hemoglobina é o que tem maior peso na hemácia
As Hemácias possuem uma grande capacidade de se deformarem, temos vasos com diamentros inferiores que as hemacias → A deformabilidade das hemácias permite-as percorrer a microcirculação sem sofrer rutura
A estrutura das hemácias altera-se em meios de diferentes tonicidades.
ao se ingerir água doce → plasmólise
ao se beber muita água, reduz muito a quantidade de sódio → assim o potencial de repouso diminui → depressão
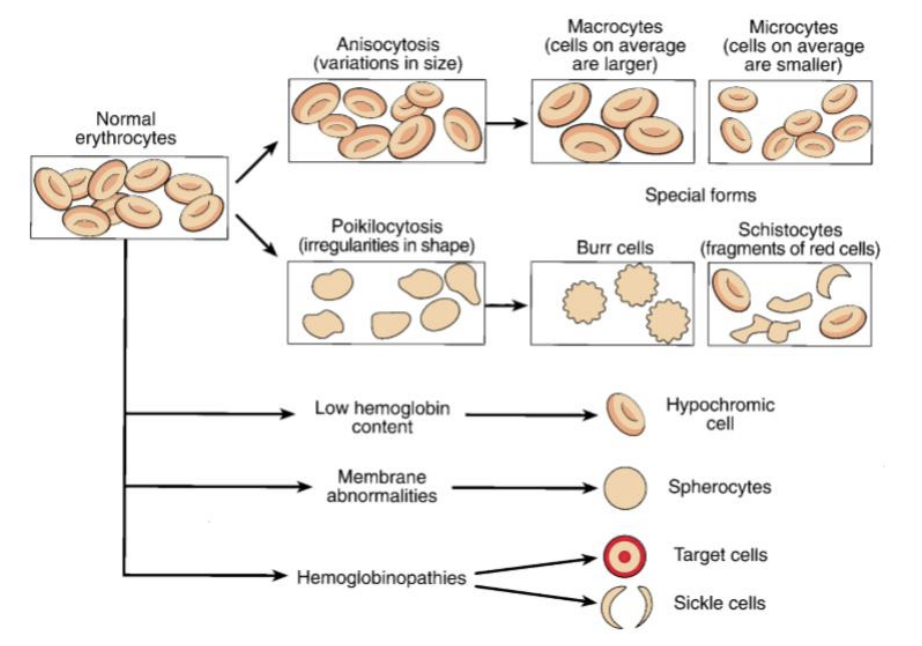
Alterações Morfológicas das Hemácias
Os orgãos que removem as hemácias:
figado, baço e medula → removem rápidamente hemácias velhas e disformes
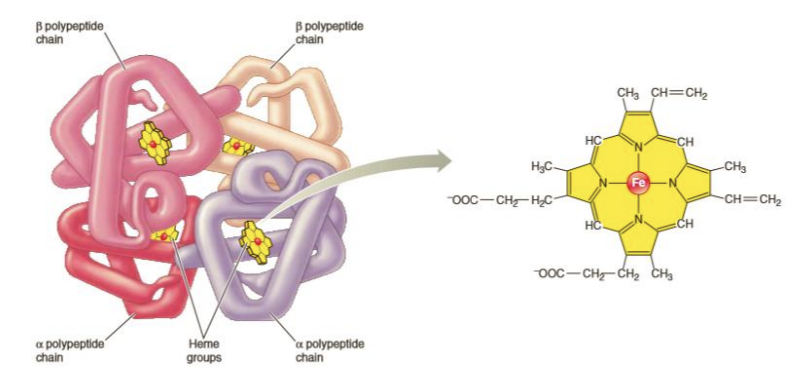
Hemoglobina
cerca de 640 milhões
Apenas Fe (II) se pode ligar ao oxigénio
Os sistemas antioxidantes da hemácia convertem Fe (III) em Fe (II)
O adulto normal contém 3 tipos de hemoglobina (Hb)
Cada tipo de hemoglobina é constituído por 4 cadeias polipeptídicas e 4 grupos heme
Uma hemácia com muito ferro III não transporta oxigénio
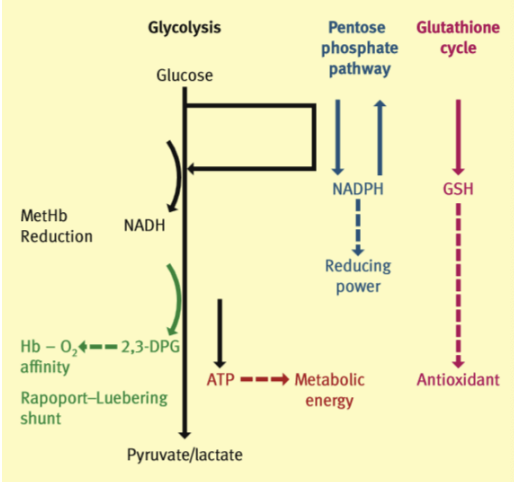
Hemácias - Metabolismo
Produzem energia por:
Glicólise:
gera ATP
gera NADH (reduzir ião férrico)
Shunt Rapoport-Luebering (mais forte)
Gera 2,3-difosfoglicerato (2,3-DPG) a partir de 1,3-DPG → via das pentoses fosfato
Via das pentoses fosfato
Gera NADPH (reduzir glutationa)
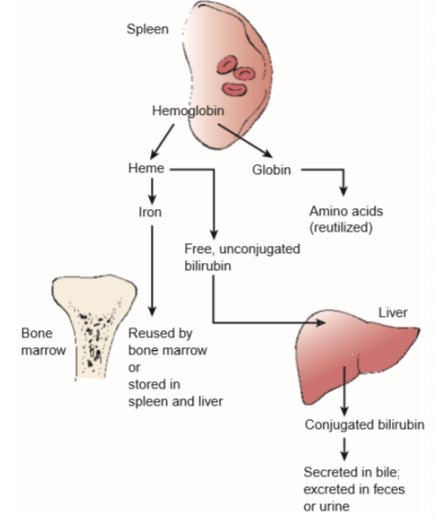
Destruição de Hemácias
Envelhecimento da hemácia
• Perda da atividade enzimática
• Diminuição da síntese de ATP
• Alteração da composição lipídica da membrana plasmática
• Aumento da rigidez celular
Destruição
• Apoptose
• Fagocitose (baço, fígado, osso)
Grupo heme – excretado na urina e fezes como pigmentos
Ferro – reutilizado na eritropoiese
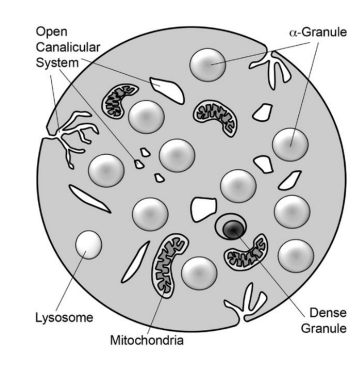
Plaquetas
Plaquetas = Trombócitos
possuem um diâmetro reduzido
Formados na medula óssea a partir da fragmentação de megacariócitos
Destruição: macrófagos tecidulares, baço
Grânulos alfa (fator de von Willebrand, fibrinogénio,…)
Corpos densos (ADP, serotonina,…)
podem estar em 2 estados: inativo (não solicitadas para o processo dehemostase) e ativas (aparencia rugosa, com pseudopodes)
Hemostase
Conjunto dos processos que tentam estancar de uma hemorragia
Hemostase = prevenção de perda de sangue
Vasoconstrição : dura minutos; pouco eficaz (transitório)
Hemostase primária = formação de um rolhão plaquetário (rolha de plaquetas)
Hemostase secundária = formação de uma rede de fibrina (proteinas)
Coágulo = rolhão plaquetário + rede de fibrina
Vasoconstrição
O traumatismo de um vaso provoca:
Contração miogénica
Libertação de autacóides por tecidos traumatizados e plaquetas (tromboxano A2 )
Reflexos nervosos (fibras sensitivas nocicetoras)
Hemorragia
= perda de sangue para fora (externa) ou para dentro do corpo (interna)
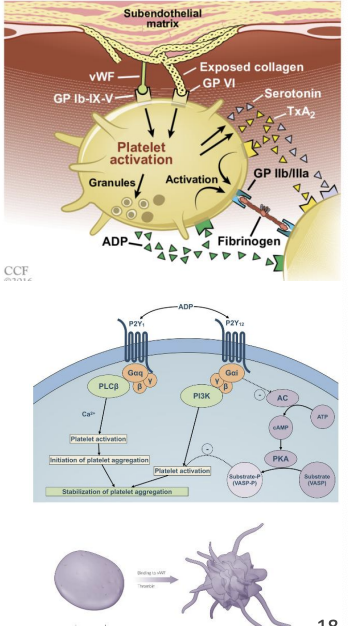
Hemostase primária
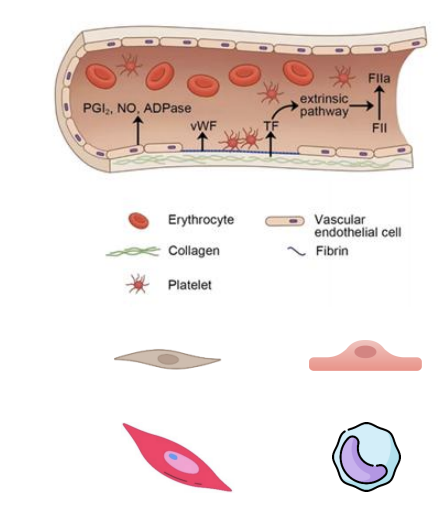
temos uma agressão a um vaso que interrompe a anatomia normal do vaso → vaso descontinuo
o que sai facilmente do vaso, aquando uma agressão, é o endotélio, portanto quando o vaso está lesado, fica à vista o sub-endotélio assim, o sangue está em contacto direto com fibroblastos, leucócitos e cél. musculares lisas → desencadeando a Hemostase Primária
Adesão plaquetária ao colagénio subendotelial (GP VI)→ ativação primária → ativação da plaqueta (proteina fica agarrada à plaqueta) → plaqueta responde ao fator (fvW) que excretou e promove a segunda ativação - ativação da glicoproteina
Adesão plaquetária ao fator de von Willebrand (GP Ib-IX-V) => ativação secundária
Secreção e ação autócrina de ADP e tromboxano A2
Ativação do recetor GP IIb/IIIa => ganho de afinidade para fibrinogénio
Protusão de pseudópodes e contração => ganho de adesividade
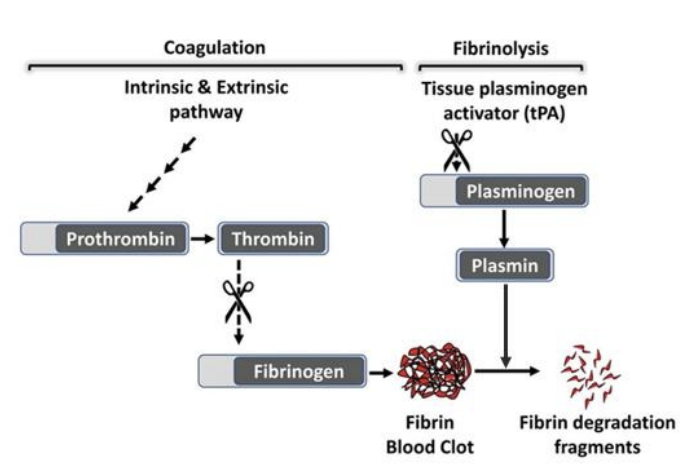
Hemostase Secundária = Coagulação
Ocorre em 3 passos:
Formação do ativador de protrombina
Formação de trombina
Formação da rede de fibrina
Queremos formar uma rede de fibrina, mas não temos fibrina, temos que a produzir → convertemos fibrinogénio em fibrina - usa-se trombina (também tem que ser produzida, apenas temos protrombina em circulação)
Fatores de Coagulação
Estão no plasma de formas inativa:
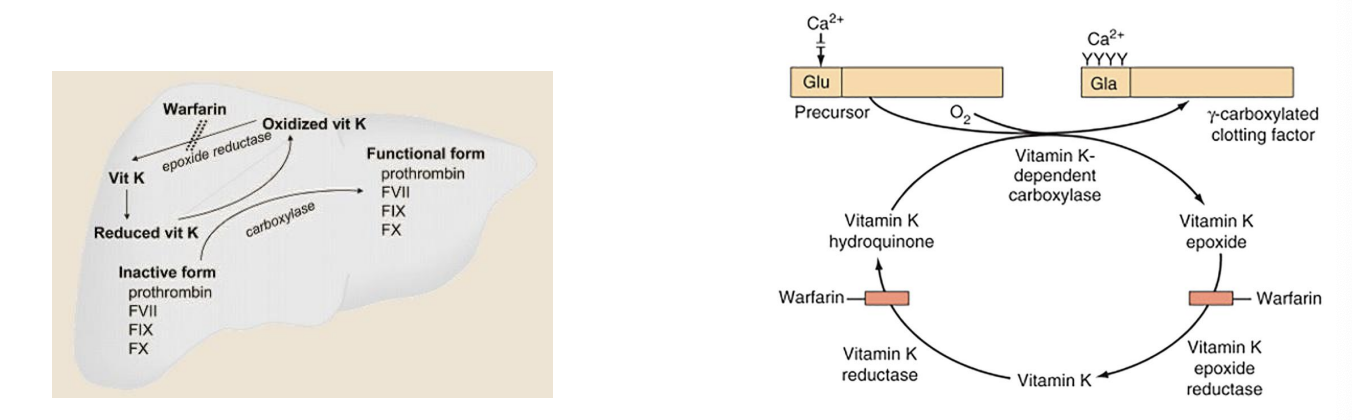
A maioria é produzida no fígado, necessitando de vitamina K
A maioria existe em duas formas – inativa e ativa (ex. fator VII (inativo) e fator VIIa(ativo))
Fator IV - cálcio - é o único que não pode estar ativo/inativo
No fígado alguns fatores de coagulação são carboxilados em resíduos de glutamato por uma enzima dependente de vitamina K (II, VII, IX, X, proteínas C e S)
Na ausência de vitamina k → fatores de coagulação inativos
Vitamina K importante para que a enzima gamacarboxilase possa carboxilar glutamato e os fatores de coagulação tornam-se passivos a ser ativos
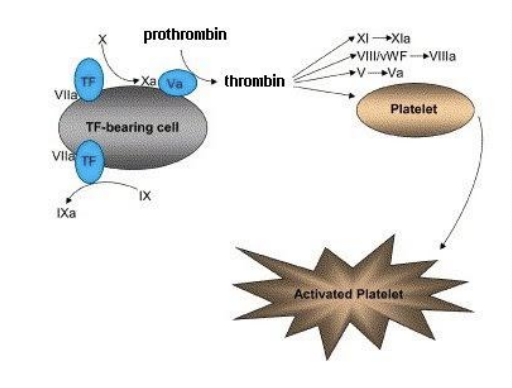
Formação do Ativador da Protrombina
O fator III com o VII juntos não ativar o fator X (7+3)
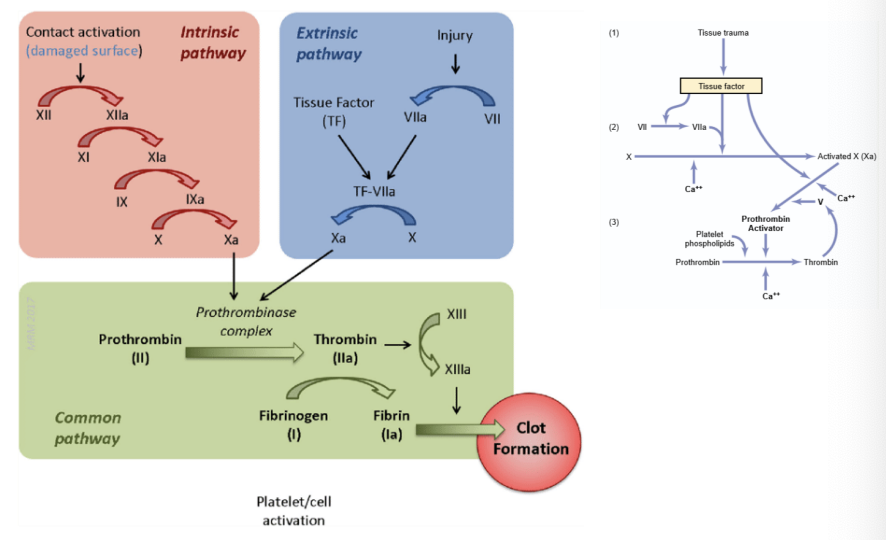
Via Extrinseca
Exposição do fator III (fator tecidular / tromboplastina tecidular) → reconhece células endoteliais, miocitos lisos e outras células
O fator III converte o fator VII em VIIa
O fator VIIa combina-se com os fatores III e IV, formando o complexo tenase
O complexo tenase converte:
O fator X em Xa
O fator IX em IXa
O fator Xa:
Converte o fator VIII em VIIIa
Combina-se com o fator V para formar o complexo protrombinase
Nota: A via extrinseca pode ativar a intrinseca; via de coagulação XIII ativado estabelece relações mais fortes
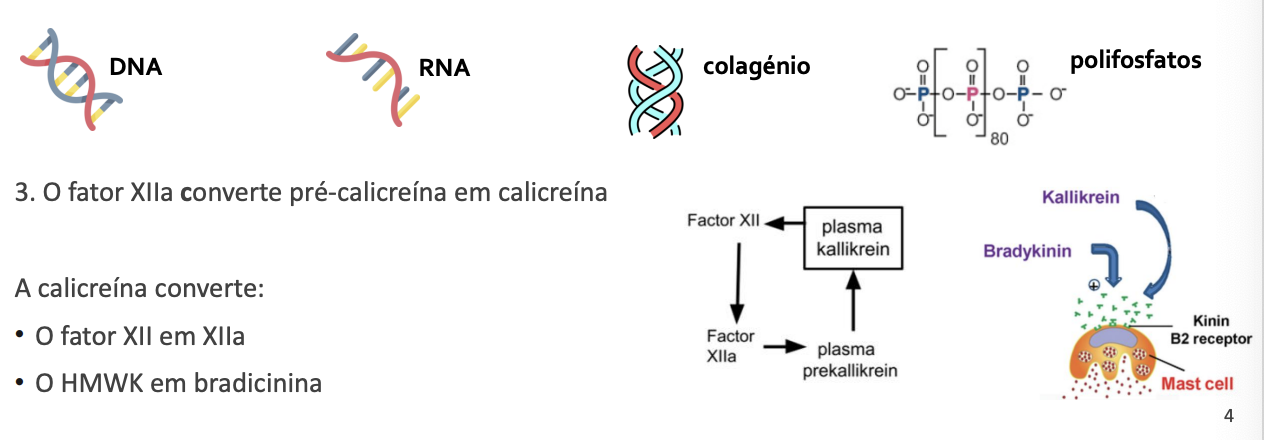
Via Intrínseca ou de contacto - ativação da calicreína
O fator XII liga-se ao cininogénio de alto peso molecular (HMWK) e forma um complexo
O complexo XII-HMWK liga-se a moléculas aniónicas no subendotélio (DNA, RNA, colagénio etc), formando-se fator XIIa
O fator XIIa converte pré-calicreína em calicreína (faz um processo de feedback positivo)
A calicreína converte:
O fator XII em XIIa
O HMWK em bradicinina (cria dor)
Via Intrínseca ou de contacto - formação do complexo protrombinase
O fator XIIa:
Na presença de HMWK converte o fator XI em XIa
O fator XIa converte o fator IX em IXa
O fator IXa combina-se com os fatores VIIIa, IV e fosfolípidos e forma o complexo tenase
O complexo tenase converte o fator X em Xa
O fator Xa associa-se ao fator Va para formar o complexo protrombinase
Nota: 12, 11, 9, 10
O fator VIII circula no sangue ligado ao fator de von Willebrand (vW) - libertado pelas plaquetas e circula no plasma
Quando o fator vW se liga aos componentes subendoteliais, desliga-se do fator VIII
A trombina converte o fator VIII em VIIIa
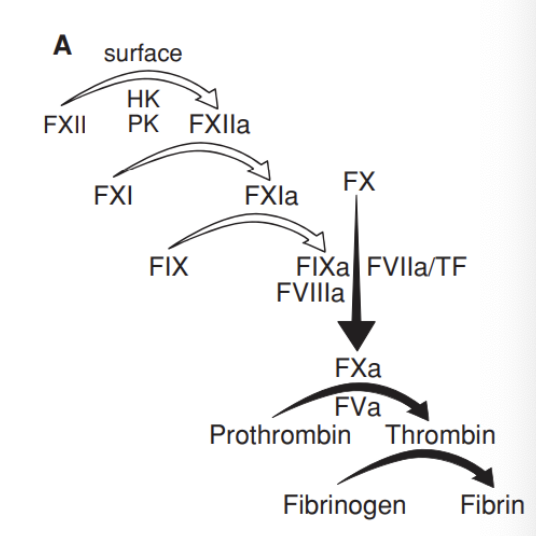
Conversão da protombina em trombina
O complexo protrombinase converte a protrombina (II) em trombina (IIa)
A trombina converte:
O fator V em Va
O fator VIII em VIIIa
O fator XI em XIa
Conversão de Fibrinogénio em Fibrina
A trombina converte:
O fibrinogénio (I) em fibrina (Ia) frouxa
O fator XIII em XIIIa, libertado pelas plaquetas
O fator XIIIa promove a criação de ligações covalentes entre as moléculas de fibrina
Nota: fator XIII - fator estabilizador da fibrina

Avaliação quantitativa da Hemostase
Tempo de hemorragia - pouco recorrido - vê o tempo de se formar o rolhão plaquetário
Hemostase primáriaT
Tempo de protrombina (PT)
Vias extrínseca e comum
Tempo de tromboplastina parcial ativado (aPTT)
Vias intrínseca e comum
Farmacos e alimentação podem alterar a tendencia a estar mais suscetivel a hemorragias
Nota: tempos estão expressos em segundos
Regulação da Hemostase
Se o sangue coagular em excesso, forma-se uma massa grande de plaquetas e fibrinas podendo fechar um vaso sanguineo, e este pode ser uma artéria, não passando mais sangue.
como frenar a coagulação:
temos 2 mecanismos anticoagulantes
destruição de coagulos já formados - fibrinólise (simples) → Plasmina corta ligações de fibrina, tonando-a mais fraca.

Anticoalogação – Antitrombina III
fator II ativado estimula a produção de fatores anticoagulantes
A trombina estimula a produção de antitrombina III A antitrombina III:
Inibe a ativação da protrombina
Inibe a ativação dos fatores IX, X, XI e XII
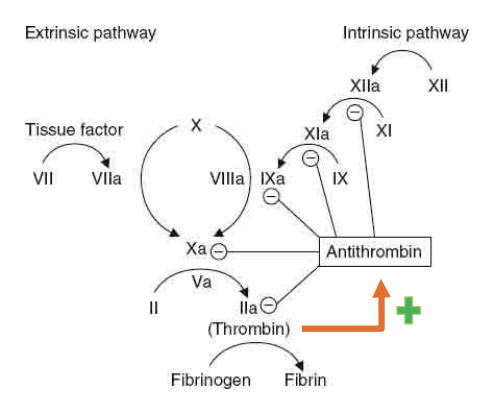
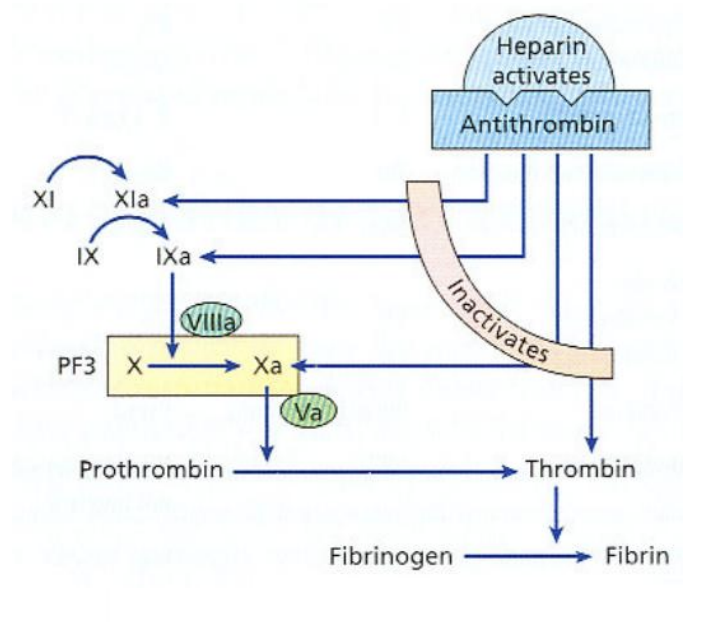
Heparina
Anticoagulante natural
Potenciador da antitrombina III
Polissacárido produzido nos basófilos e mastócitos
Liga-se à antitrombina III, aumentando a sua atividade
Inibe a ativação dos fatores IX, X, XI e XII
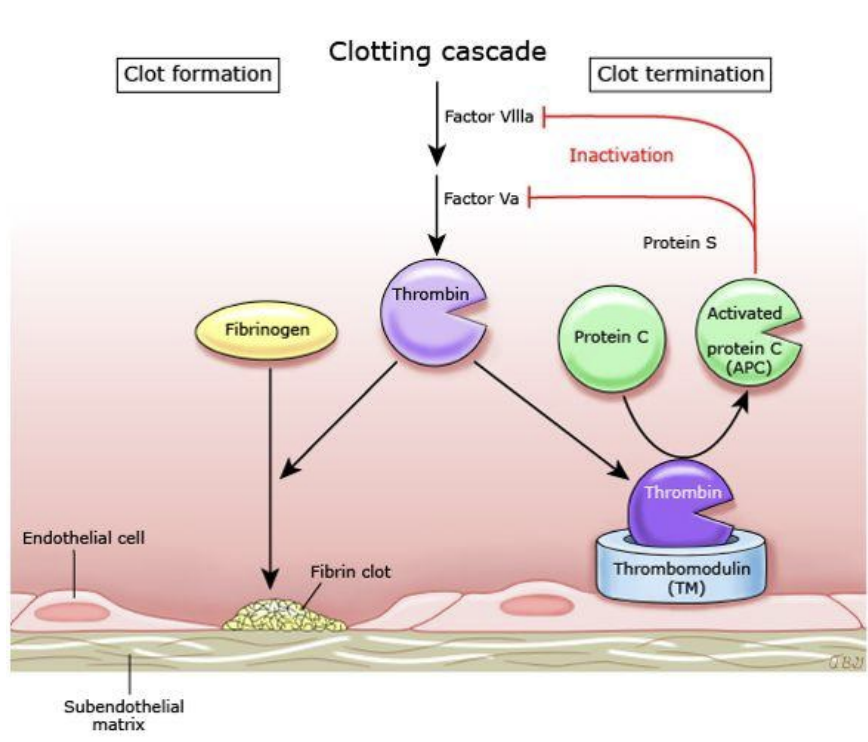
Anticoagulação – Proteinas C e S
fatores anti-coagulantes são proteinas
C não funciona sem o seu cofator, proteina S
C circula no sangue na sua forma inativa, é ativado. A partir da ligação entre o recetor trombomodulina endotelial liga-se à trombina, formando um complexo.
O complexo ativa a proteína C
A proteína C, em associação com a proteína S, inativa os fatores Va e VIIIa (diminui-se o nº de fatores pró-coagulantes)
Fibrenólise - Plasmina
coagulação
fibrinólise
Plasmina corta ligações de fibrina, está no sangue sob a forma de plasminogénio, que é ativada com ativadores, que estão no sangue ou na parede do vaso lesado.
Conversão do plasminogénio em plasmina por:
Ativador do plasminogénio tecidular (tPA) → está no vaso lesado
Ativador do plasminogénio do tipo urocinase (uPA) → circulante
Calicreína
A plasmina degrada:
Fibrina (Ia), fibrinogénio (I), fatores V, VIII, XII, protrombina (II)
Entre os produtos da degradação da fibrina encontram-se os dímeros, importantes para marcar a existencia ou não de um processo de trombose.
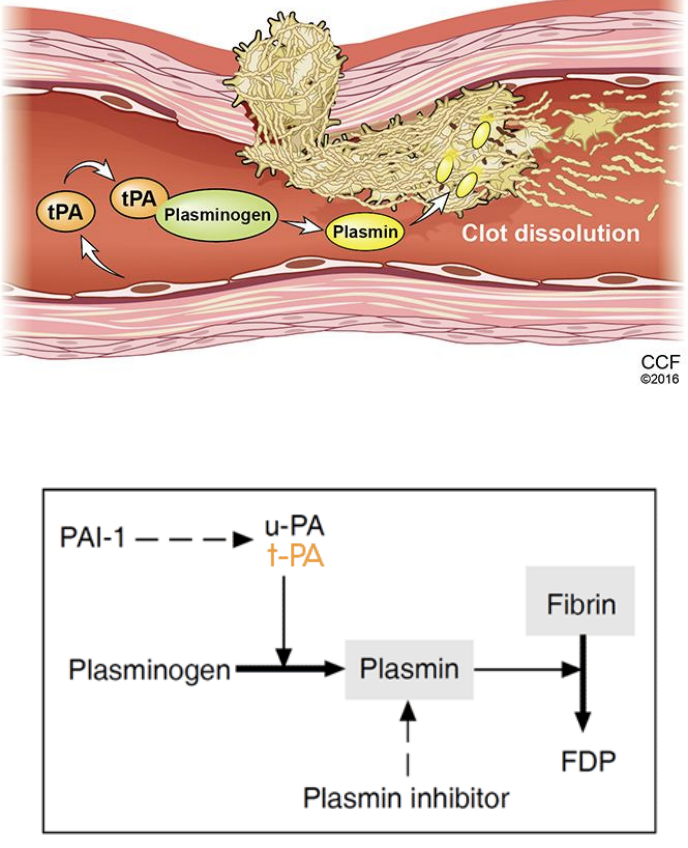
Fibronólise - Regulação
Ela própria é frenada de modo a não degradar a fibrina demasiado depressa, caso isto não aconteça, não dá para o vaso regenerar
Inibidores da fibrinólise:
Inibidores do tPA e uPA (PAI 1 e 2)
Inibidor da fibrinólise ativável pela trombina (TAPI) → complexo produz este inibidor
Desafio - Indique que risco (hemorrágico ou trombótico) apresenta um doente com:
a) Deficiente produção de fator IX
b) Deficiente produção de proteína C
c) Deficiente produção de fator de von Willebrand
trombina - converte fibrinógenio a fibrina e produz fator 13
a) hemorrágico, dado que como não tem o fator 9, não consegue reparar o vaso rápidamente
b) trombótico
c) hemorrágico
von Willebrand participa na formação do rolhão plaqietário - intervem na hemostase primária para além disso agarrase ao fator 8, e indepedo de virar 8a