Depression
1/30
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
31 Terms
Diagnosis of Psychiatric Diseases
Largely based on classification which is based on categorisation of symptoms —> describing something you have or haven’t got.
Manuals used to categorize symptoms:
DSM-5 (diagnostic Statistical Manual)
ICD-11 (international classification of Diseases)
Improved diagnosis but lacks pathophysiological definition
These diagnostic manuals don’t consider broad overlap in psychiatric conditions
As it doesn’t define pathophysiology gives no suggestions on how to treat the symptoms therapeutically
Dimensions of diagnosis
May be contribution to the disorder throughout your development —> difficult to pinpoint exactly when you’re classified as having this disorder
Psychosocial environment can also drive certain behavioural traits in some disorders such as depression and bipolar disorder.
Some clinical syndromes will differ between the prominent causing factors, E.g mental retardation has a large neurodevelopmental contribution, smaller psychosocial contribution
Genetic determinants of some disorders that are more fixed than others. Those where neurodevelopment is more crucial for the development of the disorder have a larger genetic determinant
Its not that none of these disorders have no genetic component, but some are just very difficult to pin down
No simple biological underpinning for the complex spectrum of disorders but understanding it can lead to better mitigation strategies
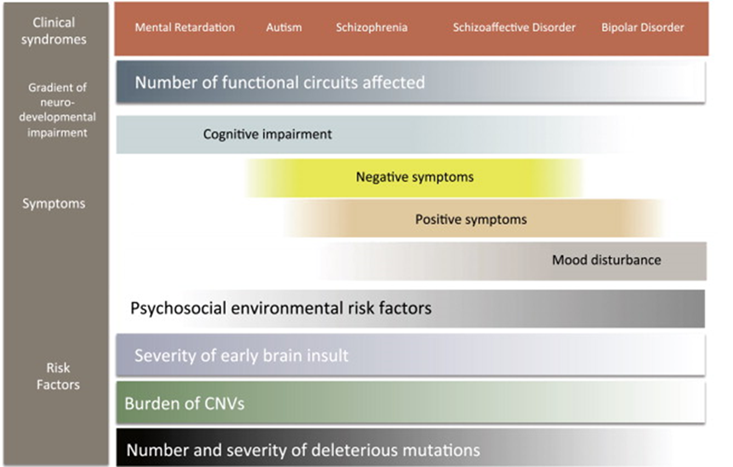
how should we treat these people - research domain criteria
Suggesting that biological interactions between genetics, environment and neurodevelopment can be used to better describe behavioural domains:
Negative valence – behaviours which apply aversiveness
Positive valence – overindulgence E.g: addiction
Cognitive systems – how you think E.g: affected in anxiety
Systems for Social Process – E.g: if youre depressed might isolate yourself
Arousal/Regulatory systems – ability to be activated appropriately affected by a condition
Sensorymotor systems – ability to move and express body language
These behavioural domains come together to better understand the psychiatric condition
the theory is that if people are distilled into categories it leads to better diagnosis
depression
pathophysiology of mood
defines as a protracted bad mood - a sustained period of bad mood where you no longer react well to the environment and it affects those around you
difficult to use inclusion exclusion criteria as its such a human condition —> diagnosis diffuse, often based on personality traits
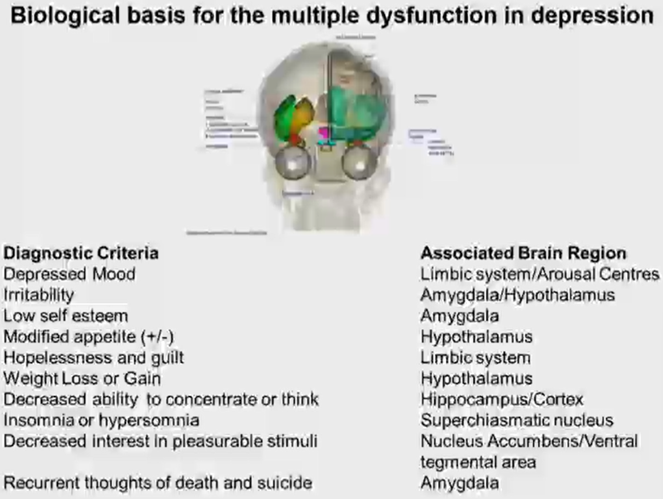
Diagnostic criteria
primary indicators:
persistant sadness or low mood
loss of interest of pleasure (anhedonia)
fatugue or low energy most days most of the time
persists for more than 2 weeks
Associated symptoms
Disturbed sleep
Poor concentration or indecisiveness
Low self-confidence
Poor or increased appetite – hypo/hyperphagic
Suicidal thoughts or acts
Agitation or slowing of movements
Guilt or self-blame
Diagnosis of disease based on primary indicators + persistence of disease + associated symptoms.
why would evolution drive us to become depressed?
Mood reflects a change in behavioural state, low mood associated with negative thought
Aversiveness is a strong reinforcer to modify behaviour associated with focus and concentration
Modifies your brain state so you can focus on something
Selective evolutionary advantage
But if you over focus on a negative event, which happens during depression, it causes a debilitative focus
however, a dysfunction in either the pathways which control focus or the modulation of these pathways will lead to a depressed state
what causes depression
a complex interplay between
genetics —> about 30-50% genetic and involved in predisposition and expression of depression
environment
sex differences —> about twice as prominent in women
defines environment genetic interaction —> genes not isolated from their environment and have a complex interaction with their biochemical and epigentic pathways
3 theories for the biological basis of depression
dysregulation of the hippocampus and Hypothalamic Pituitary Adrenal (HPA) axis
impairment of neurotrophic mechanisms
impairment of brain reward pathways
stress
depression often describes as a stress related disorder —> evidence of depression occur in some context of stress
depression in most people is caused by interactions between a genetic predisposition and some environmental factors —> “it is sensible to think not of single cause but rather of a combination of those factors that make an individual vulnerable and the external events that can trigger a depressive episode”
Dysregulation of the hippocampus and hypothalamic pituitary adrenal axis
HPA axis
HPA axis is the brains main mechanism of responding to acute and prolonged stress
neurons in the paraventricular nucleus (PVN) of teh hypothalmus secrete corticotrophin releasing factor (CRF) which stimulates the release of adrenocorticotrophin (ACTH) from the anterior pituitary
ACTH then stimulates synthesis and release of glucocorticoids (cortisol) from the adrenal cortex
this is controlled by negative feedback by the hippocampus (has an inhibiotry influence on hypothalamic CRF cotaining neurons via polysynaptic circuits) and the amygdala which exerts a direct excitatory influence
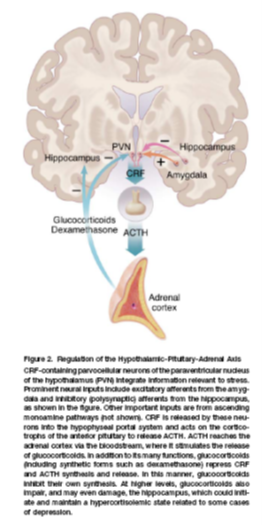
Stress’ impact on depression
sustained elevated levels of glucocorticoids (seen under prolonged stress) may damage CA3 pyrimidal (hippocampal) neurons —> reduced dendritic branching, loss of dendritic spines, loss of synapses and decreased communication between regions of the brain
elevated cortisol also causes the reduction of formation of new granule cell neurons in the adult hippocampal dentate gyrus
due to the nature of the damage of the hippocampus it prevents it from carrying out the inhibitory role in controlling cortisol levels via the HPA axis —> resulting in further increased circulating cortisol and further hippocampal damage
impaired hippocampal function may also contribute to some of the cognitive abnormalities observed in depression
Dysregulation of the hippocampus and hypothalamic pituitary adrenal axis - evidence
Evidence
abnormal activation of the HPA axis is observed in half of individuals with depression and these abnormalities can be corrected by antidepressant treatment
hypersecretion of CRF in depressed patients as seen in CSF
striking evidence between centrally administered CRF and depression symptoms: decreased appetite, increased arousal and vigilence, increased heart rate and blood pressure
limitations
unknown whether HPA axis abnormalities are a primary cause of depression or are secondary to some other cause
however, a strong case can be made for its role in generating certain symptoms and the impact of the course of disease
future treatment potentials
CRF1 receptor antagonists exert clear antidepressant like effects in several stress based rodent models
glucocortecoid receptor antagonists such as mifepristone may be useful in treating some cases of depression
impairment of neurotrophic mechanisms
Hypothesis
neurotrophic factors theory states that a deficiency in neurotrophic support may contribute to the hippocampal pathology during the development of depression
possible that antidepressant induced upregulation of BDNF could help repair some of the stress-induced damage to hippocampal neurons and protect vulnerable neurons from further damage.
antidepressant induction is at least partly mediated by CREB TF
BDNF reported to enchance synaptic plasticity in the hippocampus and so increased BDNF induced by antidepressants may promote hippocampal function and also explain why antidepressant response is delayed.
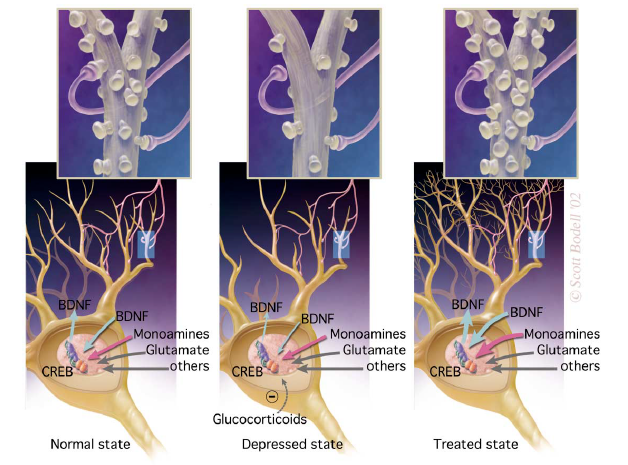
Impairment of neurotrophic mechanisms - evidence
Evidence
acute and chronic stress decreases BDNF (brain derived neurotrophic factor) expression in the dentate gyrus and pyramidal cell layer of the hippocampus in rodents
chronic (but not acute) administration of virtually all classes of antidepressants increase hippocampal BDNF levels in humans
adminstering BDNF or a related neurotrophin causes antidepressant like effects in the forced swim test
Limitations
mice lacking BDNF die shortly after birth and so cant test what happens in mice with no BDNF
since BDNF is mediated by CREB its unclear whether the increase in CREB causes the antidepressant effects or whether its the increase in BDNF
Future treatment
suggests that agents that promote BDNF may be clinically effective antidepressants
Role of CREB
Role of CREB
considerable evidence that the BDNF gene is induced by CREB
supported by the fact that all major classes of antidepressants increase CREB expression
Evidence for this
increased CREB activity in the hippocampal dentate gyrus (via viral vector encoding CREB) exerts an antidepressant like effect in the forced swim test
reason that CREB is likely to produce antidepressant effects because it upregulated cAMP. clinical observation that rolipram (a type 4 phosphodiesterase inhibitor) would be expected to increase cAMP levels and exerts antidepressant effects.
Future directions
rolipram still poorly tolerated by humans due to its side effects
but the cloning of numerous subtypes of type 4 phosphodiesterases and their specificity for regions of the brain hold promise for future development of more selective antidepressants with fewer side effects
impairment of brain reward pathways
Hypothesis
previously all research on depression has focused on the hippocampus, more recent research suggesting subcortical structures such as the amygdala, Nacc, hypothalamus have a role in causing symptoms such as regulation of motivation, sleep, apetite etc.
VTA neurons innervate the Nac as well as several other limbic structures. The Nac and its input from the VTA plays a crucial role in reward
stress causes CREB mediated transcription in the Nacc. Increased CREB in this brain region decreases an animals sensitivity to several types of aversive stimuli including anxiogenic and nociceptive stimuli. Decreased CREB in this brain region causes increased sensitivity to these stimuli. —> suggests that CREB in the Nacc controls behavioural responsiveness to emotional stimuli
the increase in CREB seen after stress of drug exposure may be responsible for the emotional numbing or anhedonia seen in some forms of depression.
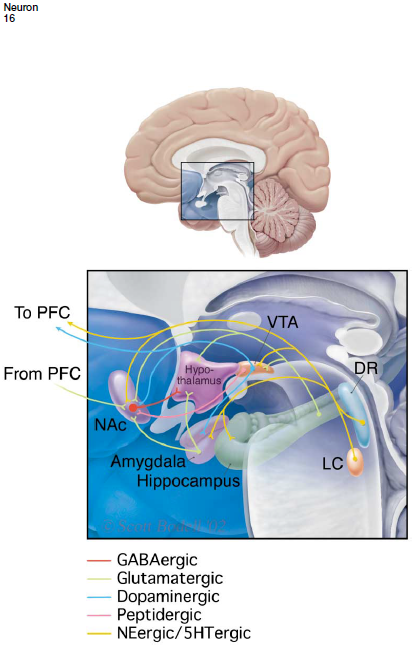
impairment of brain reward pathways - evidence
Evidence
this would explain why mice deficient in CREB show overall normal responses to antidepressants in certain behavioural tests
the amygdala (which is innervated by VTA neurons) appears to use the same cAMP and CREB pathway to promote the formation of both fear aversive and rewarding associations. Stress decreases the expression of BDNF in the amygdala.
Abnormal responses to pleasurable stimuli and symptoms of anxiety and fear are observed in depressed patients.
impact of depression on brain function
Symptomologies associated with discrete brain regions
Broad symptomology associated with diffuse and discrete parts of the brain and so underlining it is challenging
Distributed disruption of brain function —> communication across the brain is disrupted in a depression
why is depression hard to study - animal models/ learned helplessness
lack of animal models as many of the core symptoms of depression cannot be easily measured in laboratory animals
lack of known depression vulnerability genes means that genetic causes of depression cannot be replicated in animals
another weakness is that available animal models of depression utilize normal mice whereas depression probably requires a genetic vulnerability in most cases
medications required are active in the animals after acute administration while their clinical efficacy requires chronic administration
Forced swim test
a behavioural experiment we can use to measure depression in rodent models
place a rat in a tank of water and measure the amount of time it will try and climb out before it floats on its back and gives up
a reduction in immobility and an increased time trying to escape is considered an antidepressant effect.
current treatments for depression - monoamine theory
Monoamine theory
Two serendipitous observations put monoamines at the front of depression research - thought that depression is associated with an inappropriate level of neurotransmitter in the synaptic cleft – specifically biogenic amines:
Dopamine
Serotonin
Noradrenaline
Had the idea that if we were able to elevate the levels of biogenic amines in the synaptic cleft it could cure depression. There were two drugs elevated the levels of monoamines in the synaptic cleft and have been at the forefront of depression research:
Iproniazid
Trials for TB and patients experiences elevated mood
Major target was inhibition of the mitochondrial enzyme monoamine oxidase
Mitochondrial enzyme oxidises the neuroactive form of monoamines into the neuroinactive form
Inhibition increased the bioavailability of neuroactive monoamine
Imipramine
A tricyclic antidepressant
Used in trials as an antipsychotic drugs and there was an indication to improve mood
Elevated levels of monamines
Inhibits the reuptake of the biogenic amine
evidence in favour of the monoamine hypothesis
Drugs that increase/decrease biogenic amines cause increased wellbeing
Tryptophan
The starting compound for 5HT
Found in Horlicks
Increases mood
Reserpine
Prevents the reuptake of monoamines into vesicles
Causes depletion of biogenic amines
Antihypertensive with a tendency to cause depression
Measuring metabolites
Increased levels of broken down forms of biogenic amines in the CSF, consistent with a reduction of biogenic amines in depressed patients
Breakdown products include: 3-methyl-4-hydroxyphenylglycol and 5-hydroxy indoleacetic acid
Post mortem tissue
Post mortem tissue of depressed brains often have a reduction in receptors for biogenic amines
People assume that this is because of a change in genetic expression for serotonin transporter molecules but this is weakly evidenced
modulating biogenic amines
Modulating biogenic amines in the brain
Two drugs that have been used to treat depression based on their ability to modify biogenic amines and monamines
Imipramine
Tricyclic antidepression
Blocks the transport mechanism that transports biogenic amines – all have specific transporters
DAT - dopamine
SERT - serotonin
NAT – noradrenaline transporter
Has a relatively similar Kd for all of the transporters (Kd approx. for serotonin transporter is 50nm)
Fluoxatine (Prozac)
Tried to come up with a more specific transporter blocker
Fluoxetine is a selective serotonin reuptake inhibitor —> very selective for the serotonin transporter (Kd approx. 1nm)
Better selectivity in antidepressant efficacy (efficacious: produces a maximal therapeutic response)
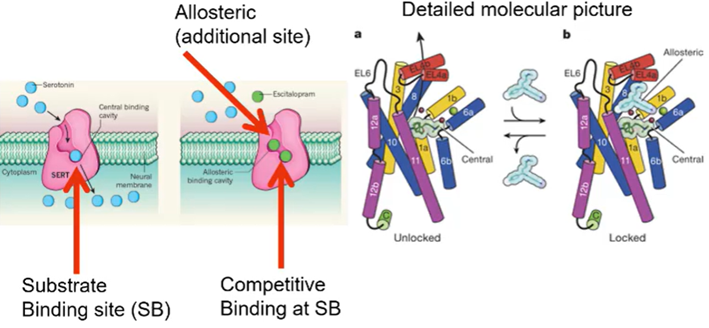
how do transporters work
Transporters are secondary transporters – use sodium and chloride, in addition to the neurotransmitter to pump from the outside to the inside
Substrate binds to the transmembrane domain
The inhibitors, the antidepressants, bind to the substrate binding site
Can bind to a second binding site on the membrane protein that carries the neurotransmitter
Transmembrane protein 1 and transmembrane protein 6 come together to form a binding site – have associated sodium and chloride binding

Escitalopram
Is a competitive inhibitor of a serotonin transporter but not only binds to the substrate binding site but also binds to an additional allosteric site
Has increased efficacy – changes extracellular loop 4 into a closed conformation which prevents the drug from leaving the active site
Supports the idea that 5Ht and biogenic amines important in expression of depression
other ways to elevate biogenic amines
NASSA drugs
NaSSA (noradrenergic and specific serotonergic antidepressants) drugs change the way which you achieve elevation of monoamine and or the receptors that the signalling is being carried by
Bind to multiple receptors which can modulate synaptic levels of biogenic amines
Neurons have biogenic amine receptors which regulate the release of themselves
Nadr is released and acting as a signal to carry the signal forward but also acting on presynaptic autoreceptos which sit on the nerve terminal. But when these receptors are activated they cause the downregulation of the Nadr release
Nadr receptors are also found on the presynaptic terminal of serotonin neighbouring neurons (no longer autoreceptors but hereoreceptors). This causes decrease in 5HT release from the 5HT containing neurons
So the same receptor can discretely regulate the levels of Nadr and 5HT
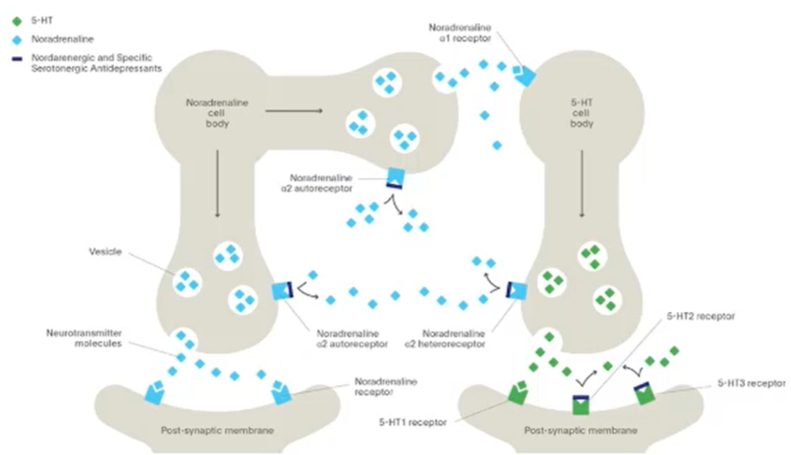
NASSA drugs block the heteroreceptos as well as the autoreceptors —> blocking a bock which causes an increase in biogenic amine release
Also act on other receptors as agonists and antagonists to modulate the sensitivity of the neurons
By binding to the 5HT 2 and 5HT 3 receptors it can reduce unwanted side effects which are mediated by these receptors such as: insomnia, anxiety, sexual dysfunction
summary of antidepressant drugs and their side effects
antidepressants need to be taken for 5 weeks to determine if theyre effective
Want to reduce side effects to increase compliance
MAO drugs – metabolise other amino acids (tyramine specifically) and so cause a lot of side effects associated with sympathetic activation
Select for one pathway with the hope that you get a better efficacy —> the associated selectivity seemed to reduce the side effects
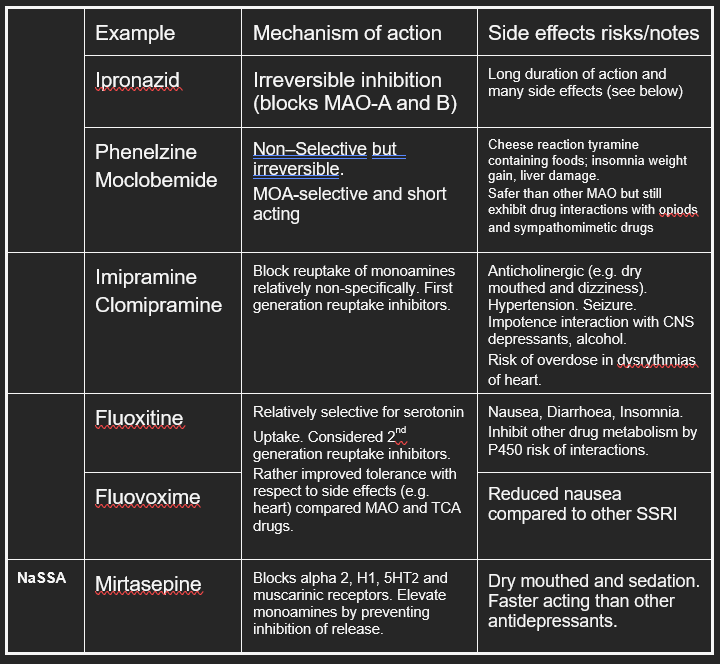
antidepressant paradox
Can see an elevation in biogenic amines in the body almost immediately but it takes 2-6 weeks to see effect on clinical signs of depression
might make patients feel worse due to the side effects but make them feel better after the antidepressant effects of the drug start to kick in
brain structure and function as drugs are taken:
Short term
Reduction in uptake causes an increased level of biogenic amine in extracellular fluid which can be measured by microdialysis
More specifically increased in the raphe nuclei, locus correlius and the cortex
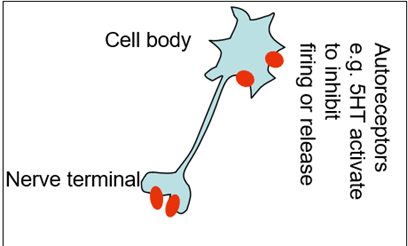
Medium term
Downregulation through autoreceptors (5ht1A receptors for example) —> does this by removal from the cell surface (a homeostatic response to maintain stable levels of biogenic amines)
Other noted changes include
Down regulation b2 postsynaptic receptors
Down regulation of a2 auto receptors
Down regulation of 5HT2 receptors.
Overall sense of a homeostatic response of pathways returns to signalling to pre-treatment levels.
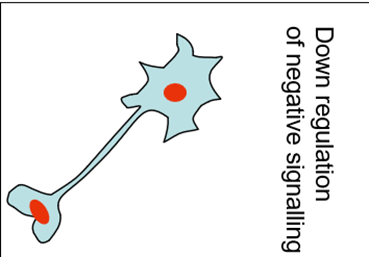
Long term
Adaptive response – plasticicity
Two main responses
Neurogenesis
Synaptogenesis
Useful for causing a significant changes in brain structure and is bought about by expression of growth factors such as BDNF
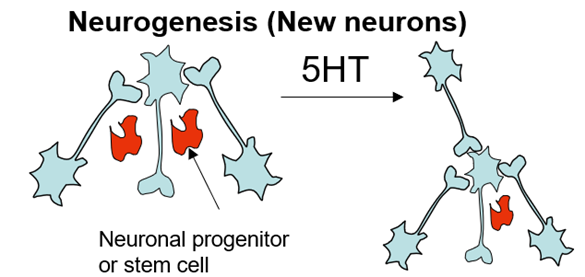
mode of action of 5HT
Mode of action of 5HT
PFC connects to all the regions of the brain associated with depression
(dorsal raphe nucleus, locus correlius, ventral tegmental area)
Aminal model used for the forced swim test but with an electrode in the brain – when you stimulate these regions do you bring about an antidepression effect?
PFC projection to the dorsal raphe – this causes release of 5Ht to many parts of the brain
Can stimulate directly at the PFC or at nerve terminals (end of the same neurons connecting to dorsal raphe nuclei)
If you stimulate directly at PFC theres no antidepressant effect but if you stimulate at nerve terminals you do
Glutamate transmitter used between PFC neurons and DR and causes serotonin release
this evidence proves that elevating teh levels of 5HTacts as an atidepresant
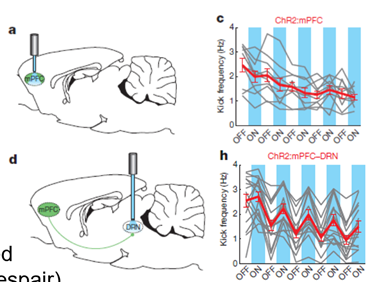

need for better understanding of cause for depression and treatment
Showed that antidepressants had a lower clinical efficacy than believed
There was no clinical efficacy relative to the placebo —> the effect of the interaction with clinicians had a positive effect on placebo group
Antidepressants had clinical efficacy for the severely depressed individuals
Clinical efficacy tested for 21 drugs including fluoxetine NASSA drugs and MAOIs and found good evidence for clinical efficacy
How likely a drugs is to be correct indicated by the LOAD score. At most this is 2-3. Biological penetrance of antidepressants in population studies is clear but the effect is modest.
You also need to consider tolerability – someone can take the drug which can have a clinical benefit but the tolerance of the drugs vary so you need to consider how tolerable the drug will be to the patient so they can take it.
In summary antidepressants have an effect that is fair but how profound the effect is and for how long it works is not well known —> there is a requirement for improvement for this as it affects so many and can be so life changing
There are drugs that we know have plasticity effects in the nervous system and needs to be explored if we can use them for the treatment of resistant individuals (ketamine being explored)
Difficulty diagnosing individuals and non-compliance can be one of the challenges with research on depression
ketamine as an antidepressant
Ketamine is a repurposed drug which is being studies as its use as an antidepressant
Quick in its action relative to SSRIs – beneficial for those which do not show immediate improvement
Acts on a different receptor to SSRIs- belongs to NMDA receptor channel blockers
Related to PCP (psychosis inducing) and MK801 (classic NMDA channel blocker)
Block a particular receptor at central glutamatergic sysnpases (excitatory synpases)
Usually these receptors rely on 2 types of ligand gated ion channels – both gated by gluatamte and lead to influx of sodium
In the case of the NMDA receptor (blocked by ketamine) it passes calcium ions which can have IC effects
Receptor has binding site for glutamate, glycine, ion binding site, channel blocking site (which works by plugging the hole, not preventing glutamate binding), NMDA binding
Produces a adose depensent response –
at 3mg/kg used for anaesthesia and works via the block of thalamic information
at 1mg/kg exhibits dissociative symptoms
at 0.5mg/kg has the possibility to be used as an antidepressant
evidence for ketamine as an atidepressant
Initial experiments to test the efficacy of ketamine as antidepressants
Double blind experiment where severely depressed individuals were first flushed out of any antidepressants they were on and tested on ketamine against other drugs that had psychoactive effects but not noted antidepressant effects. This is useful because it removes the problem of the placebo causing improvements due to the human interaction or patients knowing which drug they had been given.
Compared the treatment against these control drugs through establishment of a baseline depression using the Montgomery-Asberg depression (quantitative scoring system done by trained clinicians)
In the case of ketamine treatment there was a large significant reduction in depression score. Midazolam (a benzodiazepine used as control) showed a much less decrease in depression.
Not only was there an acute antidepressant effect but sustained effect
50% reduction in the score which is followed up by self-reporting.
About 15-20% of the patients on the trial reported dizziness and dissociation – implies that youre slightly outside the dose range, which is not good because shows that it might not be well tolerated.
Explaination for ketamines antidepressant effects
When you give ketamine to the animal, they have elevated motility in the forced swim test, indicative of an antidepressant effect
If you block this channel with a competitive inhibitor you don’t have the same antidepressant effect
Ketamine has biochemical effects in the brain —> elevates BDNF which looks as if its mediated through NMDA channel block
BDNF supports antidepressant outcomes through plasticity
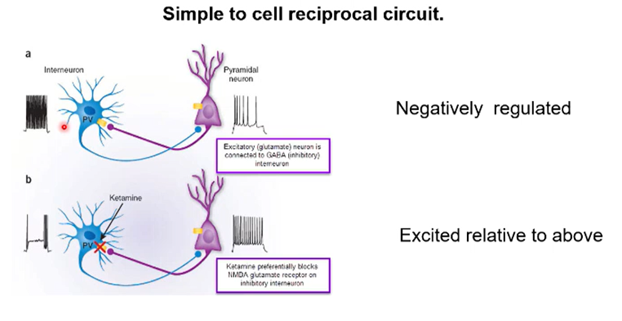
changes in underlying circuitry - theory of ketamines effect
Can model this with a simple circuit made up of an excitatory pyramidal neuron and an inhibitory interneuron
Excitatory neurons releases glutamate and inhibitory neurons which releases GABA
Inhibitory neurons excited by excitatory neurons, resulting in a reciprocal inhibition from this neuron to the excitatory neuron.
You can modify the balance between excitation and inhibition with pharmacological intervention
Drugs like ketamine selectively inhibit the glutamate receptors on the inhibitory neurons —> selectively block the excitation of an inhibitory neuron, which prevents inhibition, resulting in a super stimulation
Ketamine in a circuit could shift the balance between excitation and inhibition elevating circuit level excitation —> might explain its antidepressant effects if this occurs in the right part of the brain
other promising treatments - non drug therapies
Psilocybin
Psychoactive drug which has good potential for antidepressant, often in conjunction with CBT
Low toxicity
Acts on 5HT receptor
Allosteric modulation of serotonin signalling
Modification of synaptic function and plasticity
Deep brain stimulation
Stimulate regions of the brain with electrodes
Good change you increase circuit activity and cause acute rearrangement of brain structure which may mitigate the cause of depression
Couples with cognitive behavioural therapy
Shown to work for people with very disabling depression
Behavioual tharapy
Talking therapies offered to individuals presenting with depression
Accompanied by drug therapies
Can be looked at by brain imaging – neurochemical changes associated with it
summary/conclusions on depression
currently no definitive cause of depression - the 3 Nestler review hypotheses are some suggestions
may be one or a combination of these, which cause the biological basis, with other contributing factors such as genetics/environment.
Currently, due to a serendipitous discovery, monoamines have been placed at the forefront of treatment and research.
However, due to the fact these only show moderate efficacy with chronic administration (despite the fact biogenic amine levels are increased immediately) it suggests that increasing biogenic amine levels in the synaptic cleft does not produce antidepressant effects. Rather it is the long term plasticity and change in brain structure that produces the antidepressant effects shown by these drugs, and therefore supports the view of the Nestler theories that there is a deeper underlying cause affecting multiple brain structures.
Research on depression is difficult and has setbacks due to
difficulty diagnosing patients
multiple factors at play causing depression
limitations of animal models
varying severity of depression
difficult to quantify symptoms
tolerabiliity of drugs impeding research