Organic Chemistry - Test #3
1/52
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
53 Terms
Thermodynamic Favorability
Thermodynamics: The most stable product will be formed in a favored reaction
Favored reactions release energy
Exothermic: reactions that release heat vs. Endothermic: reactions that require heat (enthalpy H)
Exergonic: release energy vs. Endergonic: reactions that require energy (free energy G)
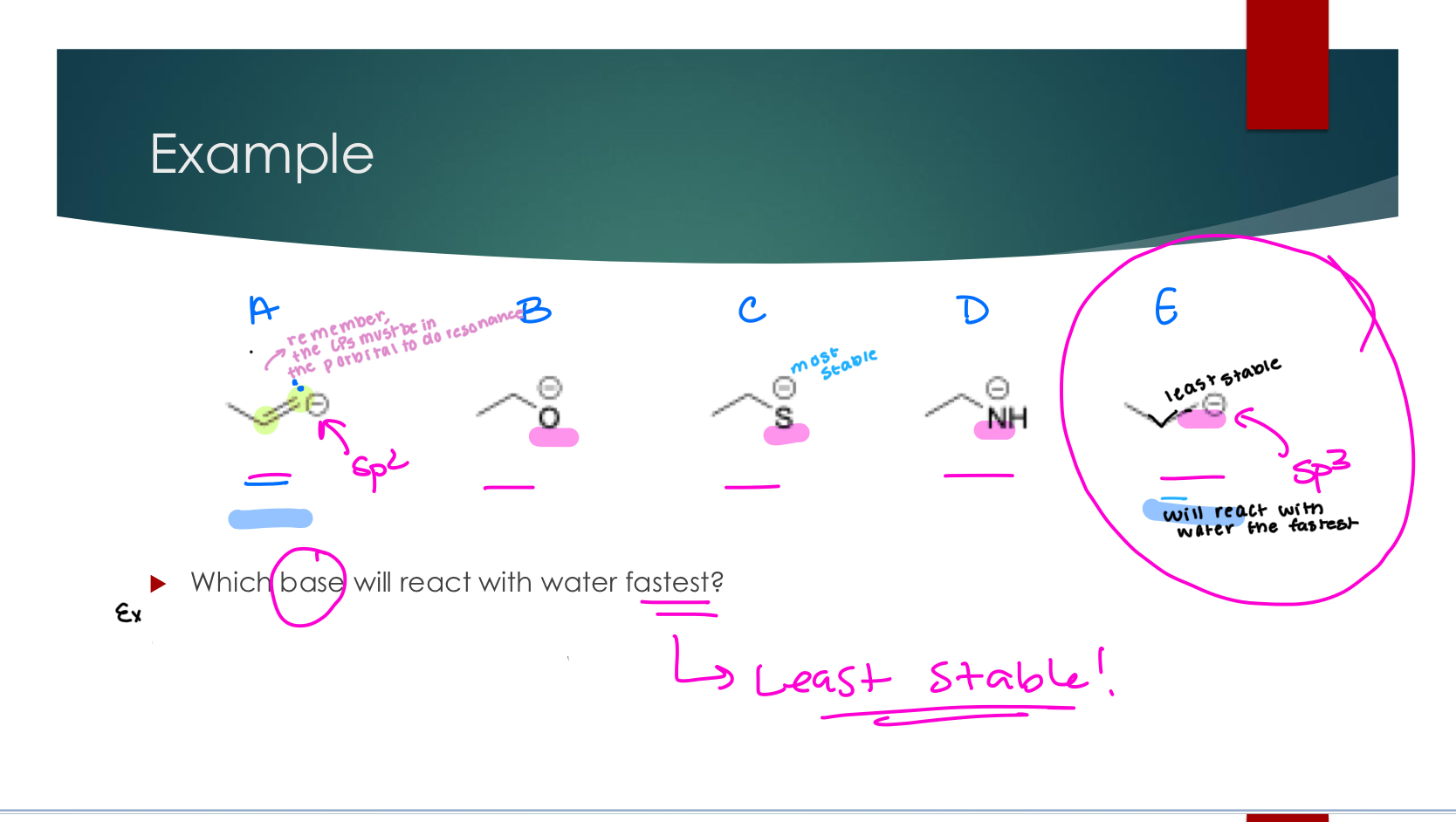
To assess which direction a reaction is thermodynamically favored: compare the stability of both sides (focus on the most unstable compound on each side... Charges!)

Kinetic Favorability
Kinetics: What will happen fastest?
The more unstable the reactant the faster it will react
The easier the transition state is to access, the faster it will react
FAST DOES NOT EQUAL FAVORABLE
To assess which reaction will occur faster: compare the reactivities of the reactants or the activation energies / energy of the transition states
General Principle: Reactions that only involve the transfer of a single proton (Acid/Basen Reactions) are kinetically very fast!
> Practical Take Away: if an Acid/Base is possible, it will happen first.
Collision Theory & Molecularity
Molecules have to physically collide with each other in order to react, so the things that affect how quickly product can be made are:
Number of collisions = possession time: if one team has the puck 90 % of the time, you’d guess that they’re have a higher score.
Required Orientation of Collisions = shots on goal, you need the right shot to score, you can shoot all you want but it needs to go in.
Required Energy of Collisions (Activation Energy or Ea) = Goalie: low activation energy it’s a bad goalie, and you’re going to get more goals. High activation energy and it’s a good goalie, and you’re going to get less goals.
Statistics: How probable for molecules to collide → Reaction Molecularity
Unimolecular: The reaction happens within the same molecule, requires no collisions with another molecule
Bimolecular: The reaction requires two molecules colliding
Termolecular: The reaction requires three molecules colliding
Mechanisms & Elementary Steps
Reactions typically occur in a series of elementary steps : description of what molecules collide to lead to a product
Typically unimolecular or bimolecular
Overall reaction: Summary of all reactants & final products
A reaction mechanism is the order of elementary steps that must occur to generate the final product
Intermediates: chemical compounds produced in one elementary step and used in another elementary step, these compounds only appear in a mechanism, not an overall reaction
Mechanisms are just a guess of how things happen, and they’re ot factual this is how this happens.

Elementary Steps & Rate Laws
A rate law shows the relationship between reactant concentrations & reaction rate
Zero Order: the rate doesn’t depend on that reactant’s concentration
First Order: the rate has direct relationship to the that reactant’s concentration
Second Order: the rate has an exponential (square) relationship to that reactant’s concentration
Rate laws can be written for elementary steps by using the reaction stoichiometry using the format:
Rate = k[reactant in step]stoichiometric coefficient of that reactant
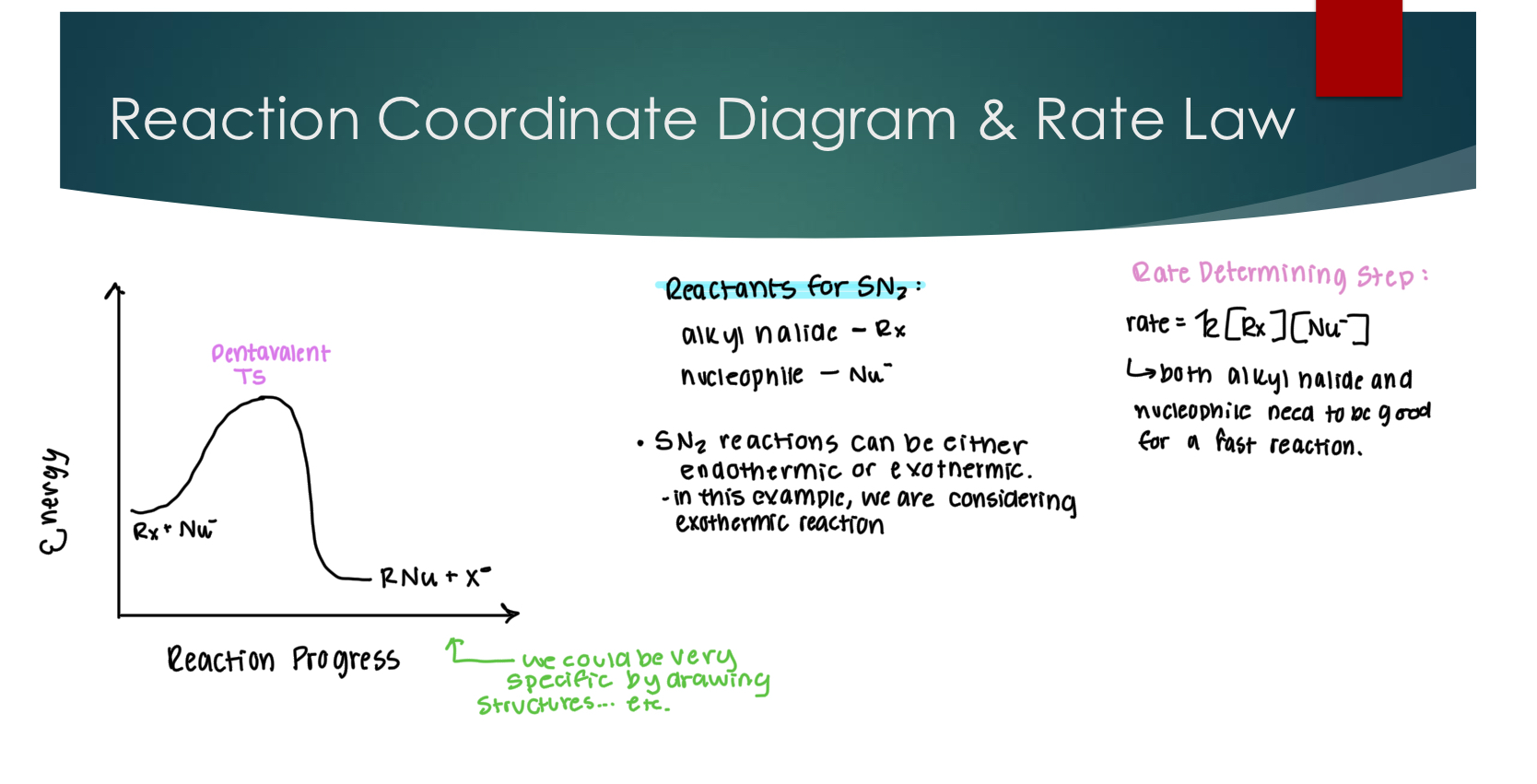
The rate of the overall reaction is dependent on the slowest elementary step, called the rate determining step (RDS)
Overall rate law = the rate law of the RDS
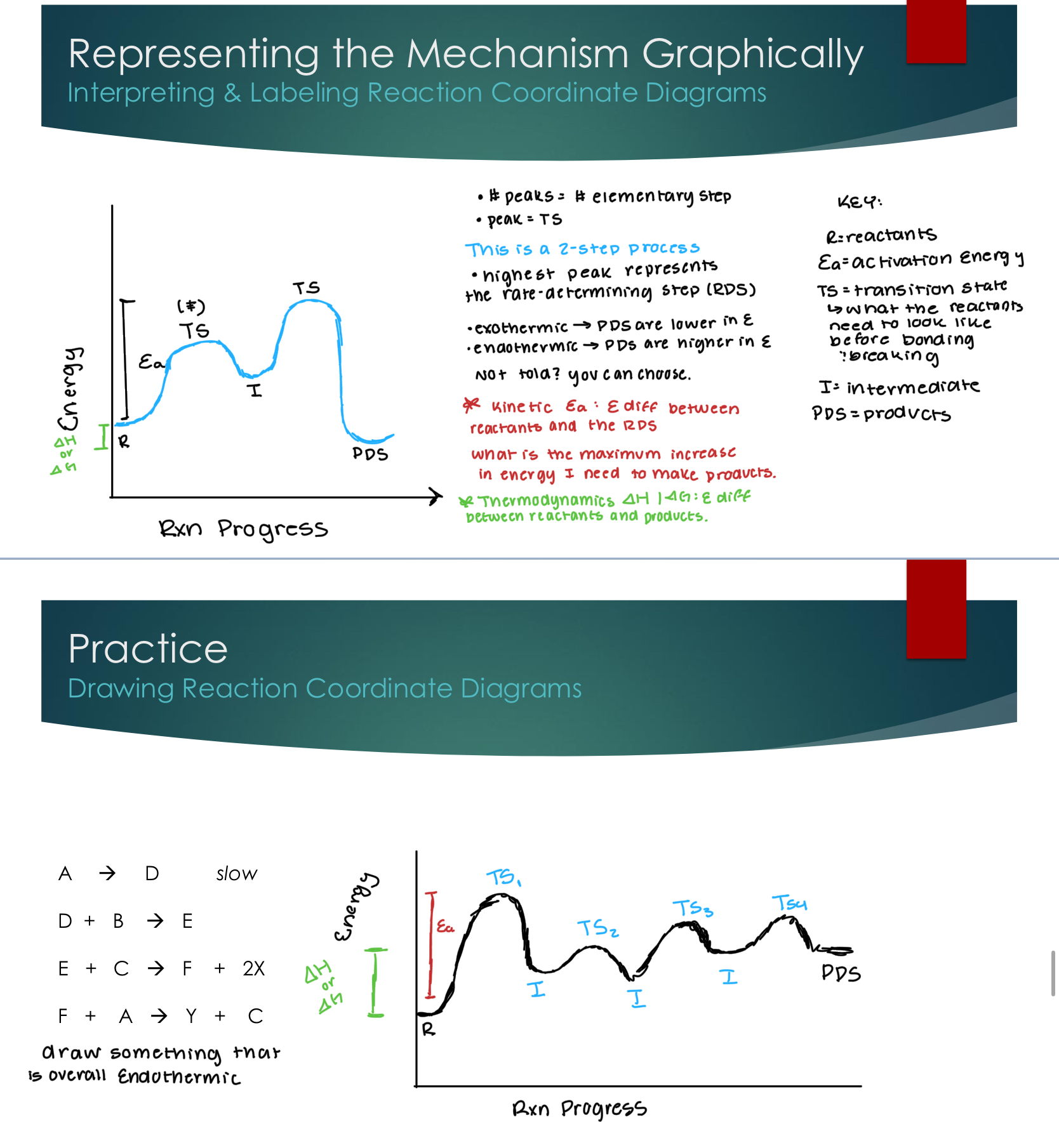
Representing the Mechanism Graphically
Interpreting & Labeling Reaction Coordinate Diagrams
The number of peaks is the number of elementary steps
The peak is the transition state
The highest peak represents the rate-determining step
Energy levels can be determined
Exothermic → PDS are lower in energy
Endothermic → Products are higher in energy
Kinetic Activation energy is the energy difference between the reactants and products.
What is the maximum increase in energy I need to make products.
Thermodynamics ∆H/∆G: energy difference between reactants and products.
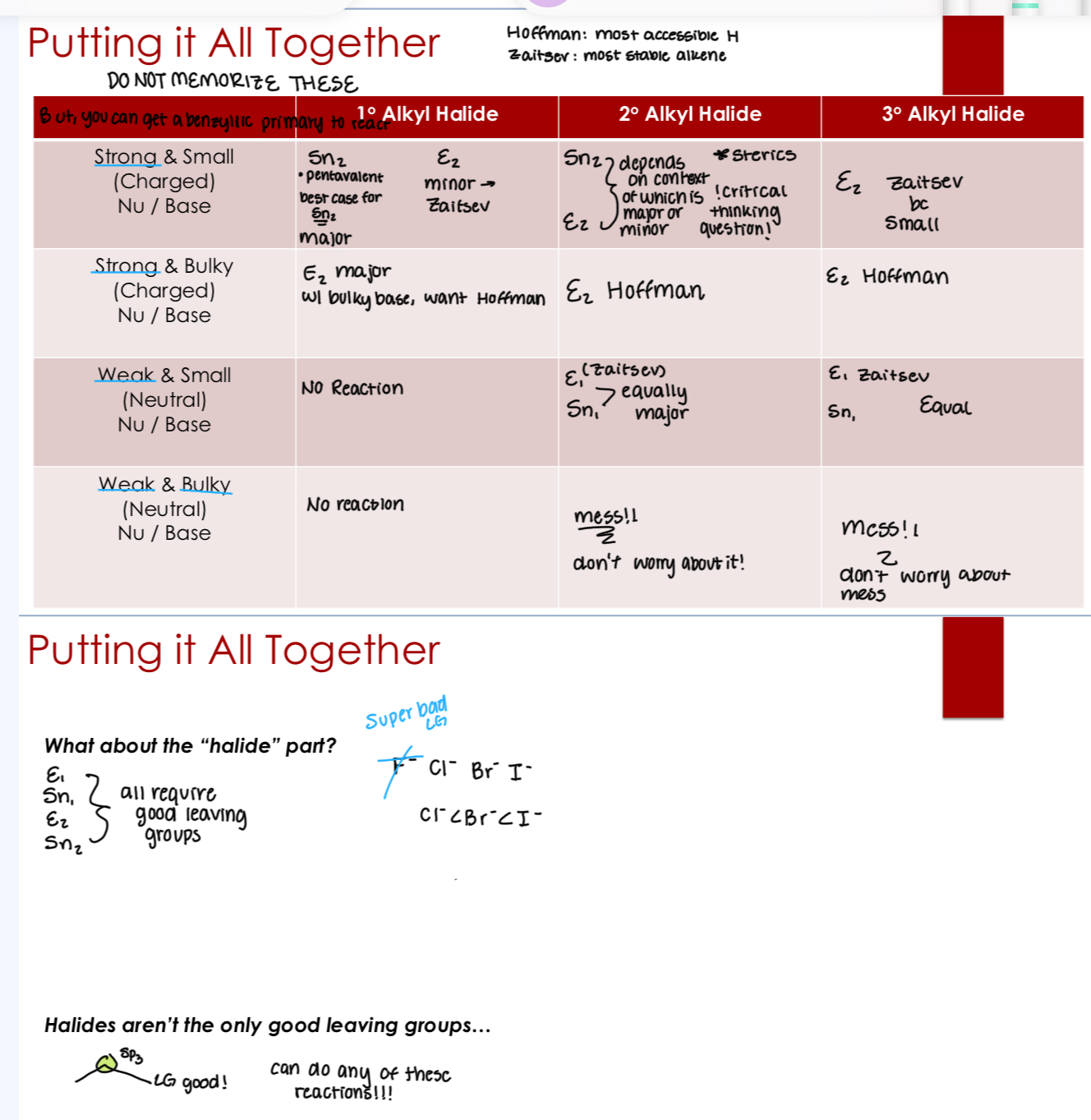
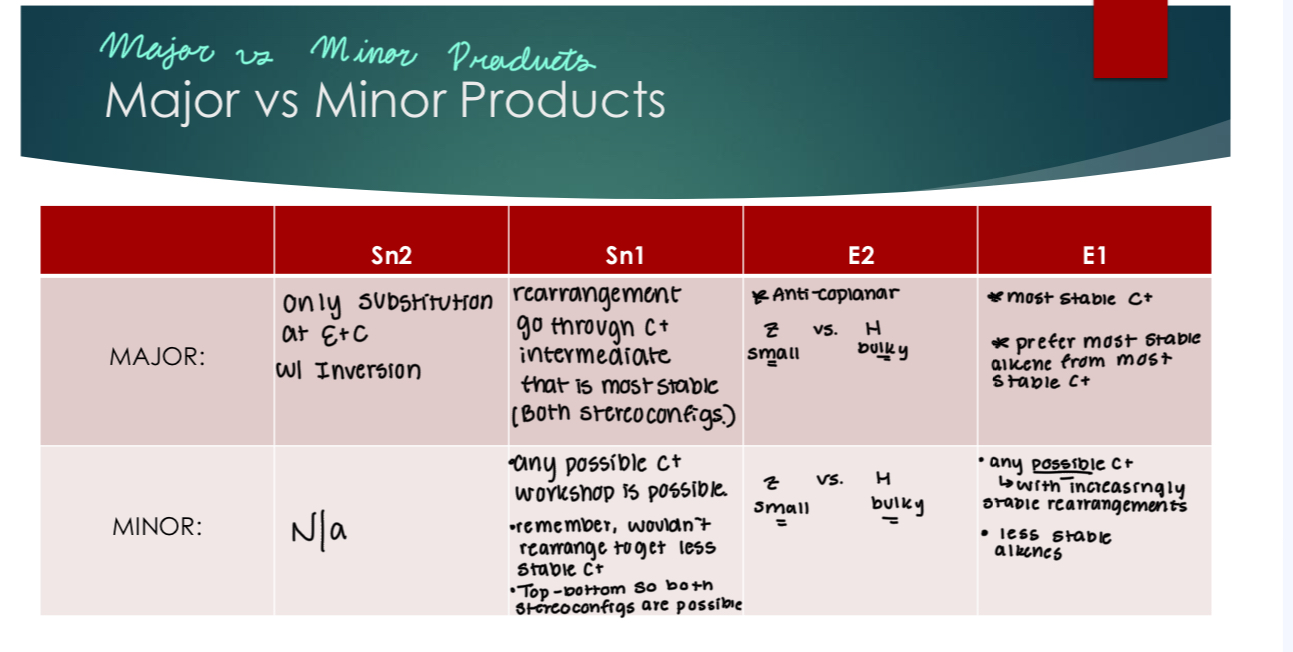
Major & Minor Products
Some reactions can lead to multiple products
Major product(s): the product(s) that is/are formed in greatest quantity (whatever you make more of)
Minor product(s): the product(s) that is/are formed in lesser quantity
When there’s one product, there’s only one major and no minor
Major products are typically:
More stable (thermodynamics control the products)
Formed by the fastest mechanistic option (kinetics control the products)
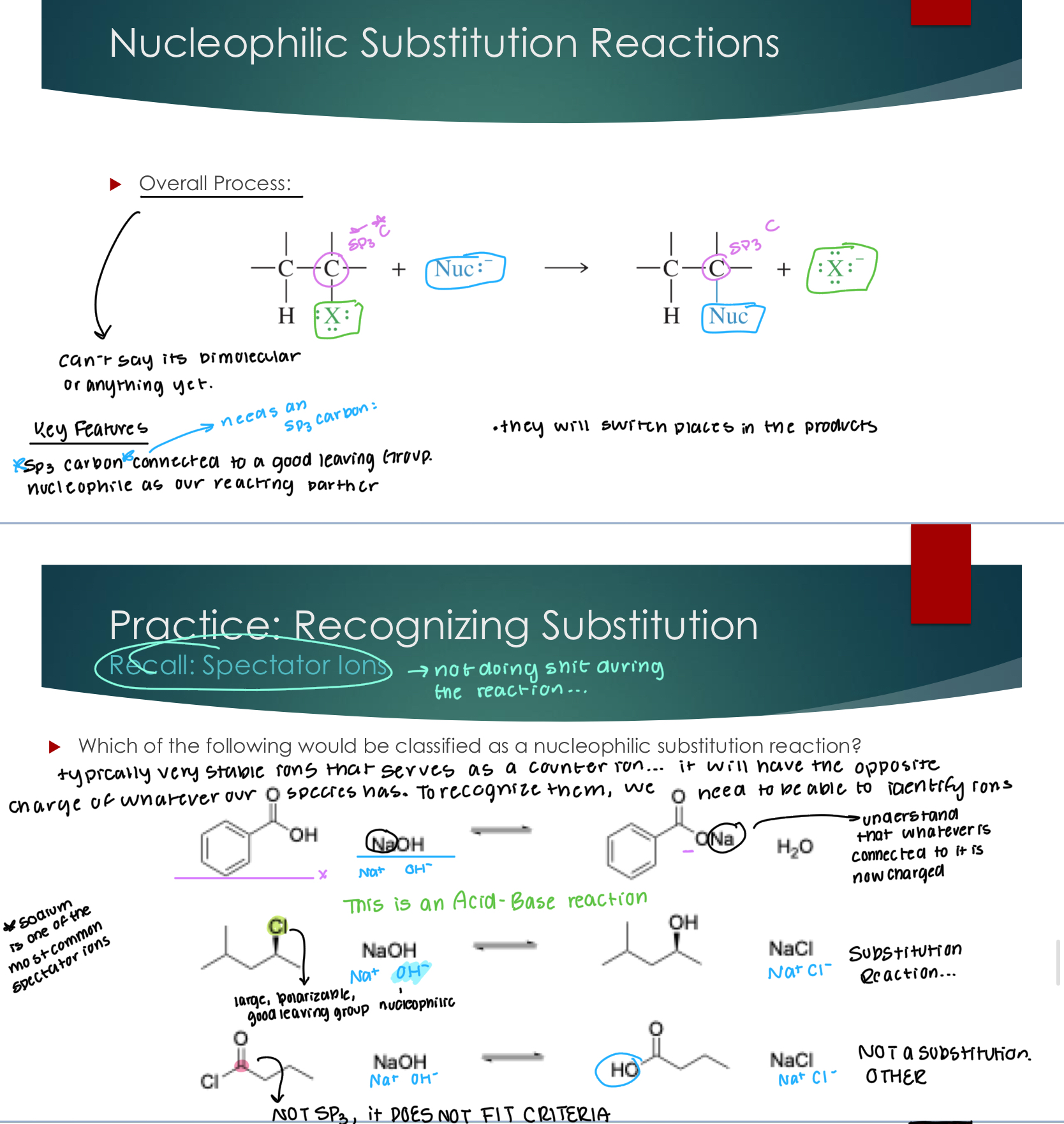
Nucleophilic Substitution Reactions
Can’t say it’s bimolecular or anything because we don’t have the elementary steps.
KEY FEATURES:
SP3 Carbon is connected to a good Leaving Group. N
Nucleophile as our reacting partner
They will switch places in the products…
Typically, very stable ions serve as a counter ion… it will have the opposite charge of whatever our species had. To recognize them, we need to be able to identify ions.
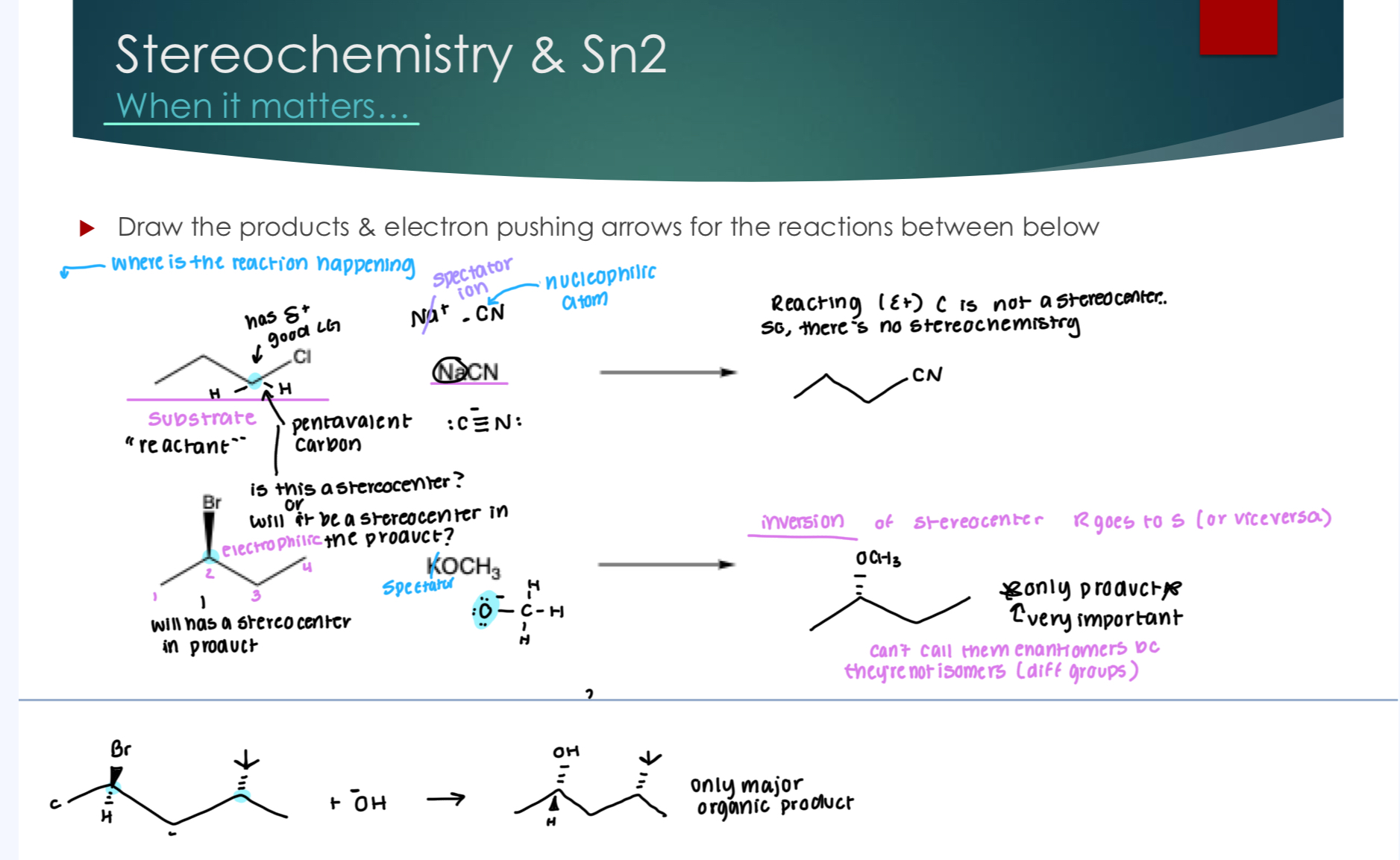
The Sn2 Mechanism: The first mechanistic possibility for substitution
KEY FEATURES
Only one step
Only one transition state
Concerted: bonds forming and breaking at the same time
Two things are colliding in the same step: bimolecular
RDS is our only step.
When can this happen? What will the collision look like?
Orientation must be correct: 108° orientation where nucleophile is attacking and leaving group is leaving.
Nucleophile must do backside attack to generate a transition state
Pentavalent: 5 things around carbon
smaller groups easier to put around C, which leads to a better reaction.
We have to thing about pentavalent rules to see if this is eligible to happen.
Want to overlap for the backside attack to maintain the 180° orientation (look at orbital diagram if you don’t understand why).

Reaction Coordinate Diagram and Rate law for SN2
Stereochemistry & Sn2
Stereocenters in the products have inversions, where R would go to S and S would go to R. Note, it’s only the product and specifically the stereocenter affected that has the inversion, not the other stereocenters within the molecule.
Good Nucleophiles for Sn2
Want something that isn’t bulky!! Long chains of carbon are NOT bulky: there are general rules (like primary, secondary, and tertiary) as seen by the chart, but you don’t want bulkiness because then the nucleophile can’t reach the halogen to be able to do the backside attack.

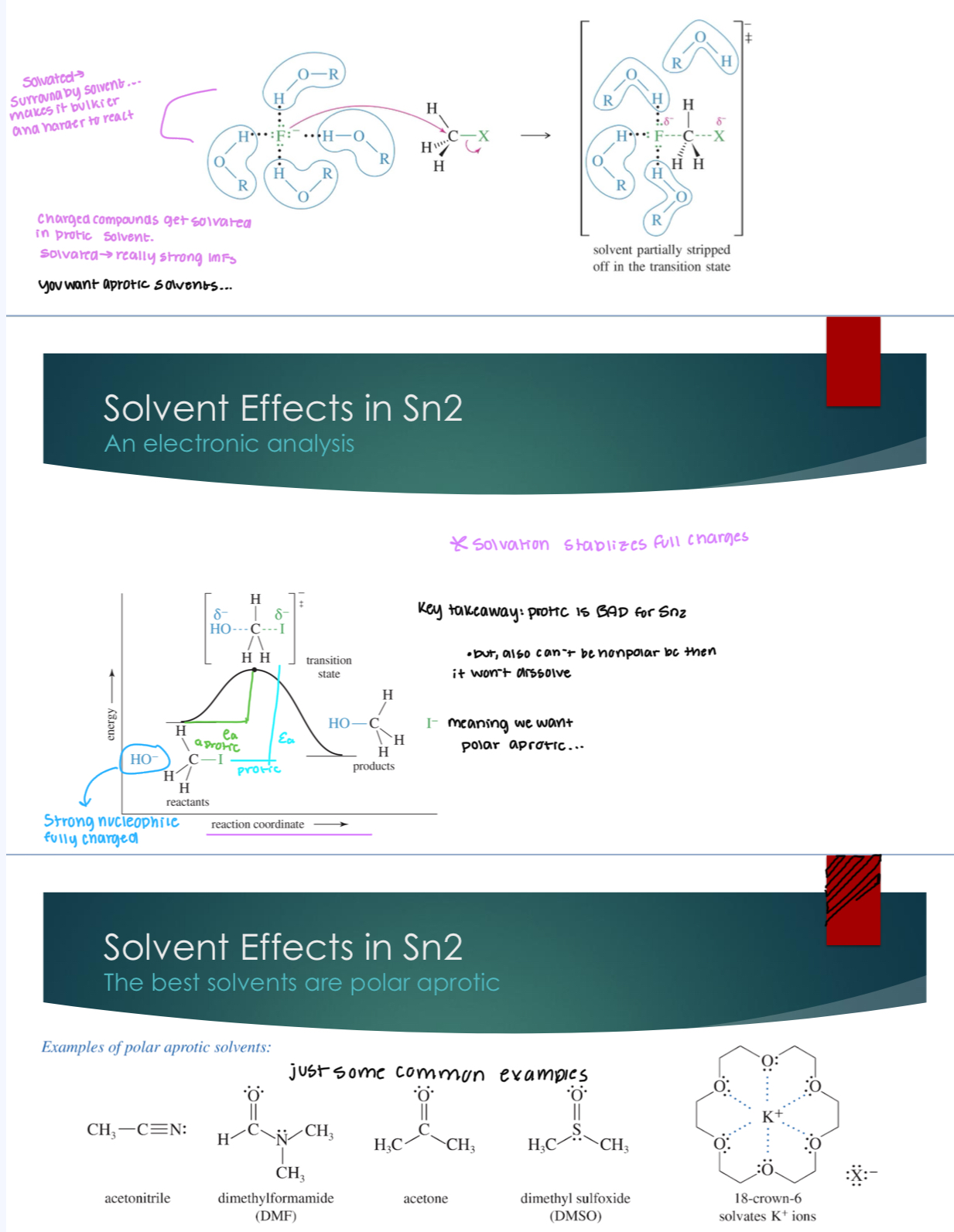
Solvent effects in Sn2
protic solvent: is when there is at least on H-bond donor.
aprotic solvent: when there is a solvent with no H-bond donors.
protic is solvated (surrounded by solvent) which makes it bulkier and harder to react. Charged compounds get solvated in protic solvent so we want aprotic solvents but NOT aprotic solvents.
An Sn2 reaction is still possible in a protic solvent, it will just be slower
➢ Solvent effects are the last priority when considering how “good” an Sn2 reaction will be, and in determining if a different mechanism will proceed instead.
➢ In general, the solvent will not be the only reason a reaction will not go through a Sn2 mechanism
Sn2 Summary

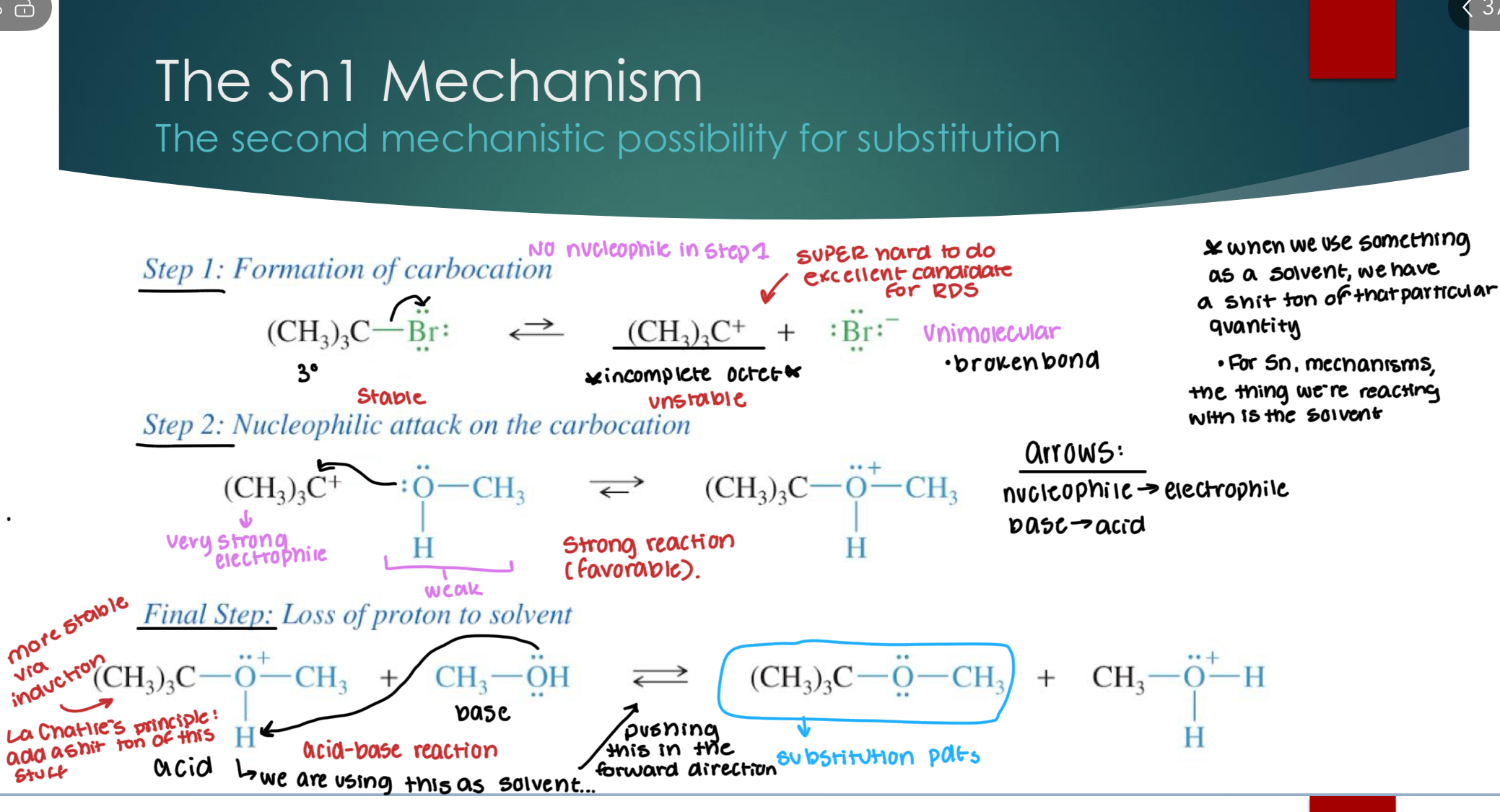
The Sn 1 mechanism
*When we use something as a solvent, we have a shit ton of that particular quantity. For Sn1 mechanisms, the thing we’re reacting with is the solvent.
Step 1: Formation of the carbocation (unimolecular)
Step 2: Nucleophilic attack on the carbocation
Final step: loss of proton to solvent, (deprotonation)
The RDS in this case is step number 1 (formation of the carbocation) because you are taking a very stable reactant and making a very stable product. Plus, Acid-Base reactions happen extremely quickly, and step 2 is very favorable and therefore very fast.
For this reaction, we are focusing on anything that makes Step 1 better and faster, and easier (which I will discuss). Because it will make RDS faster.
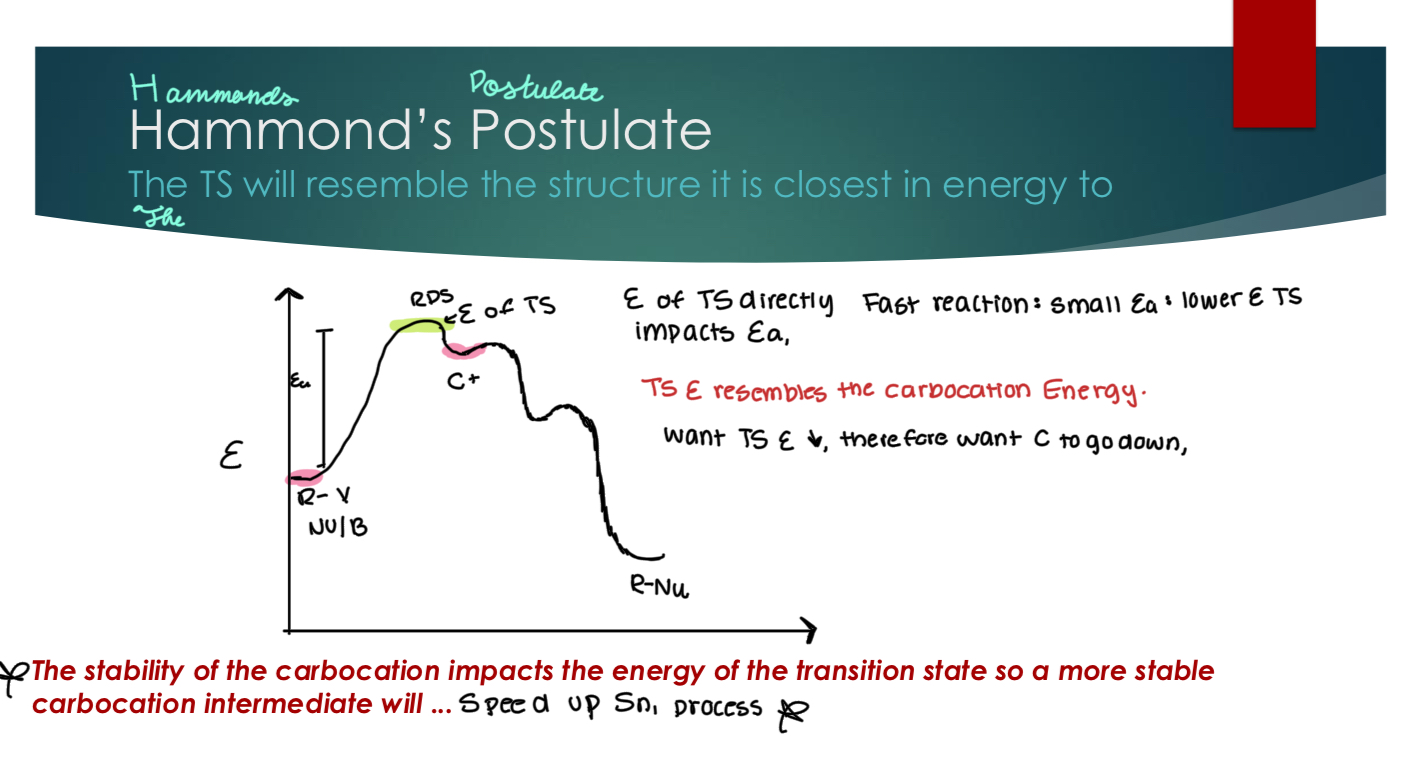
Hammond’s Postulate
It states that the transition state will resemble the structure of its closest to in energy. So, E of the TS is close to the C+, and the E resembles the carbocation energy. Which means, we need to reduce the energy for the carbocation (by making it more stable) to reduce the activation energy for the RDS. In other words, The stability of the carbocation impacts the energy of the transition state so a more stable carbocation intermediate will speed up the Sn1 process.
The Carbocation Intermediate
The key aspect of the Sn1 mechanism is the carbocation intermediate!
Carbocations are inherently unstable
Anything that stabilizes the carbocation will make the reaction better (Hammond’s Postulate)
Carbocations will do whatever they can to get more stable: Rearrangements
Carbocation are sp2 so their geometry is: trigonal planar AKA flat!!
Can an sp2 carbon be a stereocenter? NO
The nucleophilic addition to the carbocation controls the stereochemistry of the product(s)
For the stability of the carbocation, we want a tertiary alkyl halide and NOT a primary alkyl halide. (to be a good electrophile).
Nucleophiles in Sn1
Does the nucleophile impact the rate of the Sn1 reaction? Nope! Not in the rate law.
The nucleophile reacts with the carbocation, so does it need to be strong for this step to be favorable? We already have a strong carbocation, so we need a weak nucleophile/base for this to work.
Typical nucleophiles for Sn1 are therefore: solvent quantity (and is possible when it’s weak)
When reacting with solvent: (solvolysis) a synonym for Sn1 type process
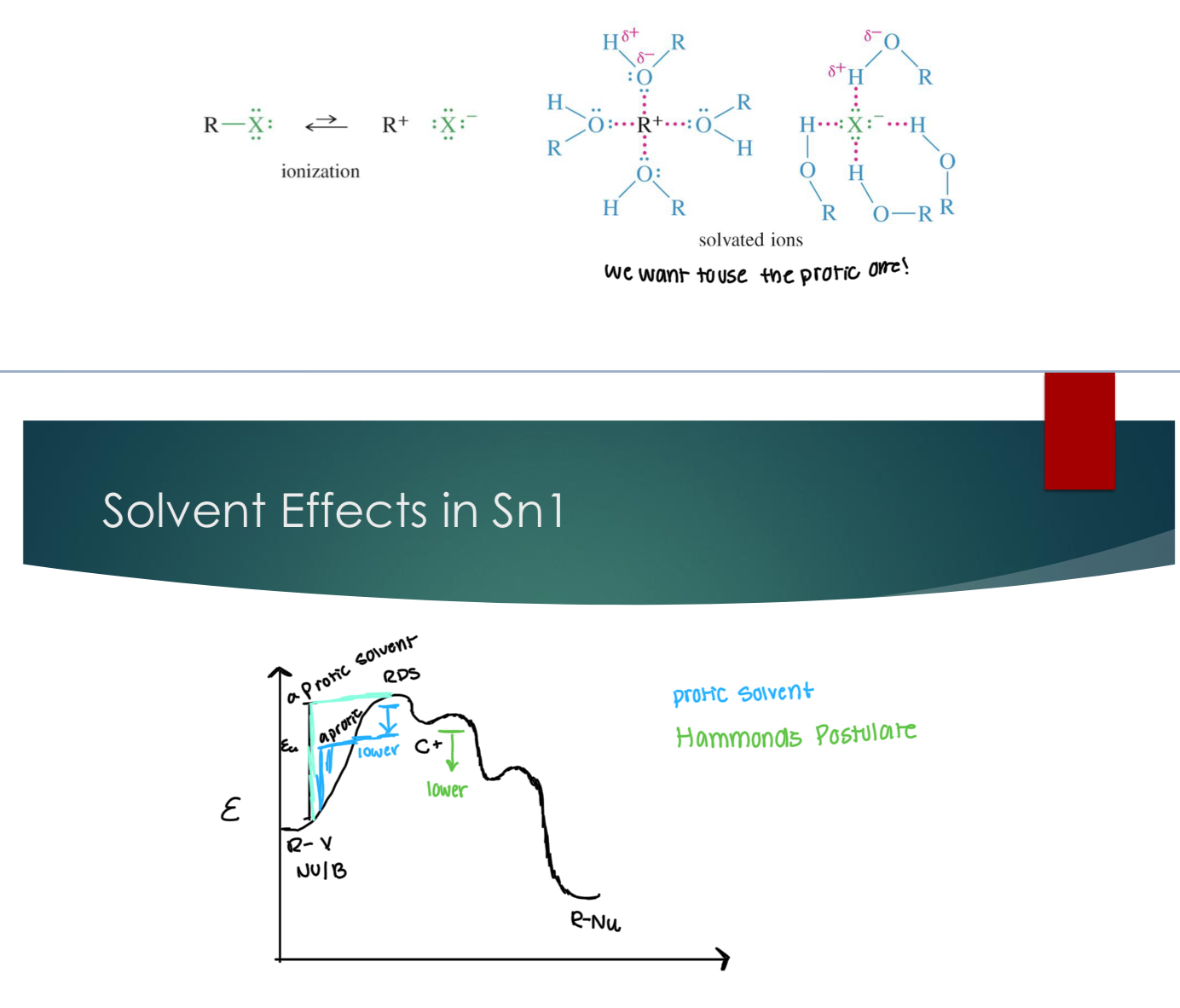
Solvent Effects in Sn1
Protic: H-bond donors → stabilize charges via solvation. We want to stablize the carbocation. So we want to use protic solvents!!
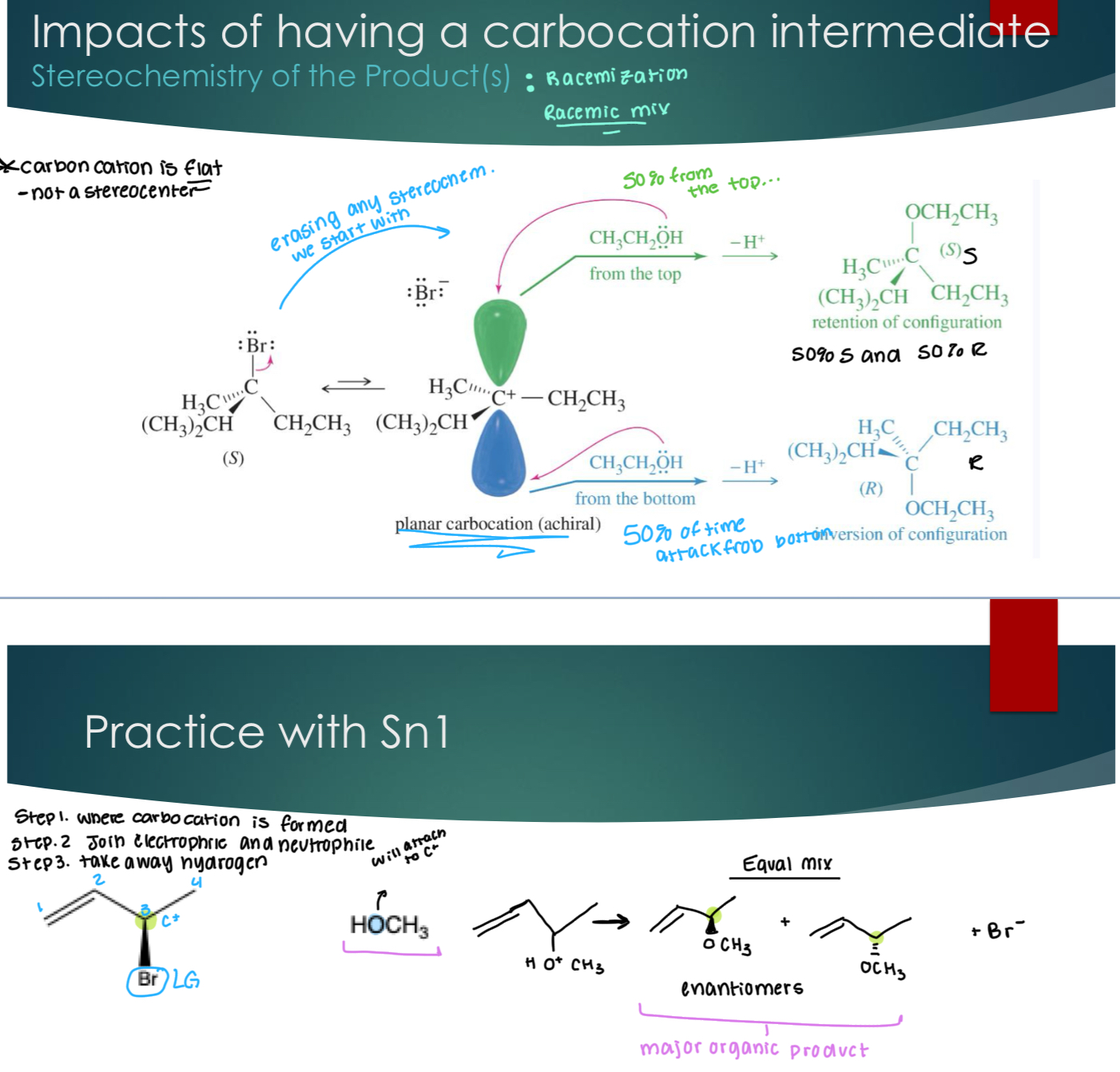
Impacts of having a carbocation intermediate
Carboncation is flat, NOT a stereocenter
So, you get two products when the original molecule has a stereocenter because it doesn’t matter when you approach it from the top or the bottom.
Do NOT memorize that Sn1 gives you enantiomers, it can also give you diastereomers if there are more than one stereocenter.
Sn1 Summary

Carbocations are really unstable... If they can easily rearrange to increase stability, they will!
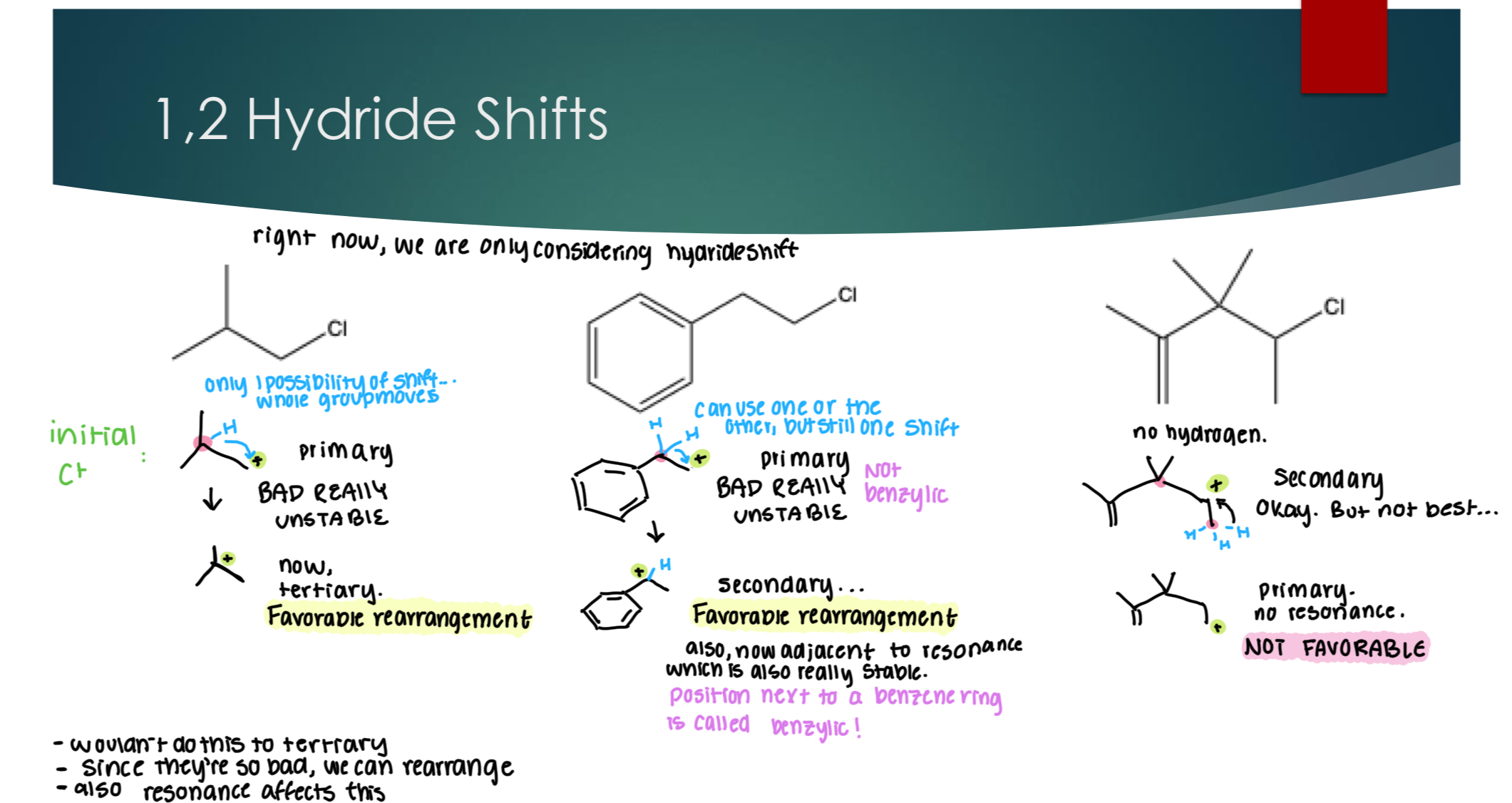
1,2 – Hydride Shift (H w/ two electrons): relation between C+ group and the group that is shifting. Has to be in an ADJACENT position.
1,2 – Methyl Shift: H3C- w/ two electrons in bond.
Carbocation shifts only occur when: it results in a more stable carbocation.
In general, when both types of shifts can produce equally stable carbocations: Hydride is major!
1,2 – Hydride Shift
Things to note:
Carbocations who are already tertiary will not do this
Since primaries are so bad, we can rearrange
Resonance does affect this.
A position next to a benzene ring is called benzylic.
1,2 – Methyl Shift
Evaluating if the methyl shift is better…
Note: if the C+ is next to an alkene, its called allyic. FYI: primary, secondary, and tertiary are called substitution!.
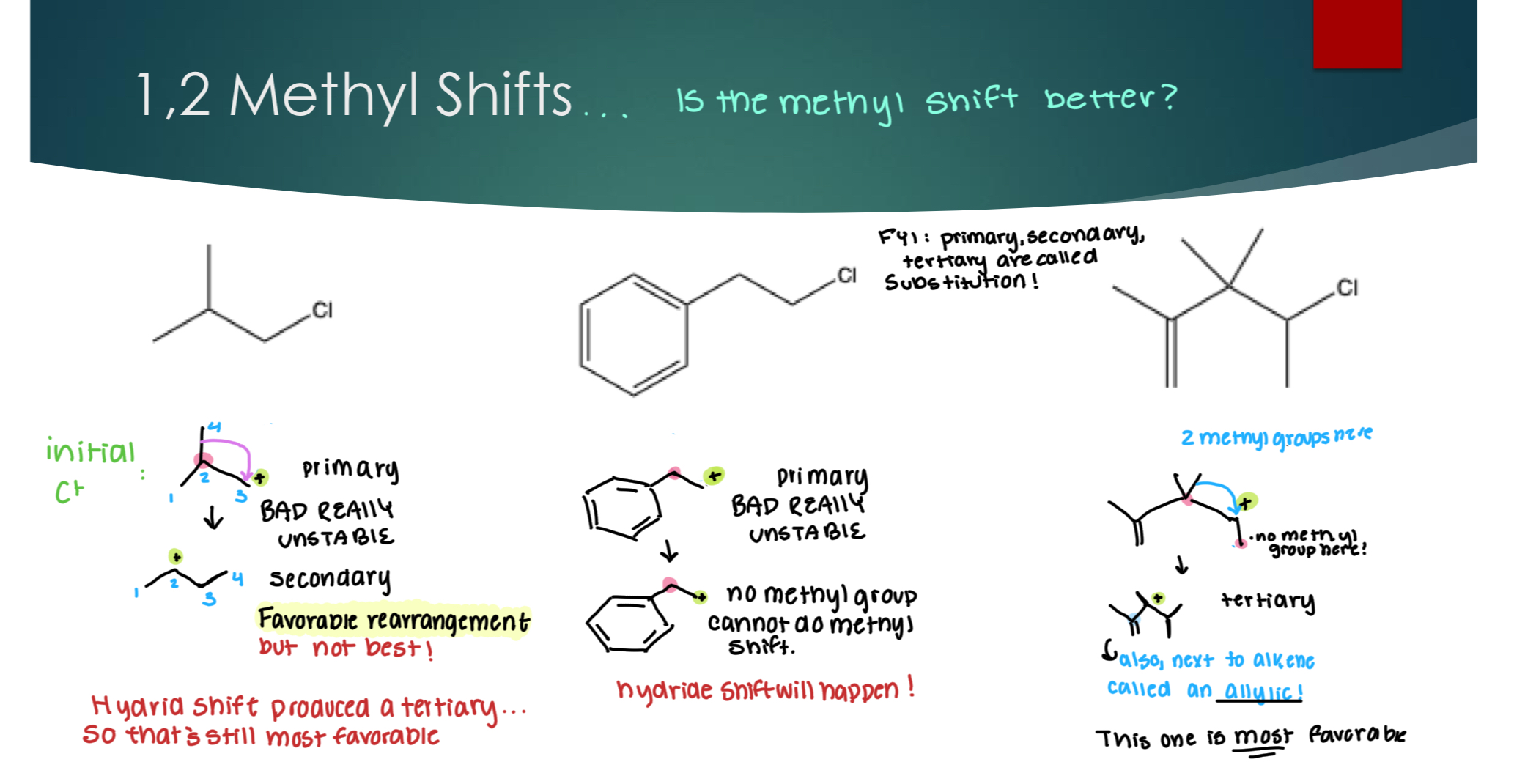
Selectivity Concerns when evaluating rearrangements:
Major is the best organic product, while minor is one of the following conditions:
addition to initial C+
When a rearrangement is favorable, but not the best.
Other Selectivity concerns:
Many nucleophiles can also act as bases and bases attack protons only:
Meaning that you’d want a partial + hydrogen as seen in the example.

Elimination Reaction introduction
Can occur through multiple mechanisms, and like substitution are named
based on the molecularity of the rate determining step:
E1: unimolecular elimination (1 molecule in RDS)
E2: bimolecular elimination (2 molecules in RDS)
Generate alkenes as a product: next functional group focus!
The E1 Mechanism: What are the similarities to the Sn1 mechanism?
Starts with a molecule undergoing a reaction.
Elimination uses a base to attack an acidic hydrogen and then uses that to create a double bond between the two carbons.
Kinetics of the E1
What is the RDS & rate law? Step 1 (from carbonation) rate = k[Rx]
What kind of alkyl halides will be best for E1? Tertiary and secondary alkyl halides, as well as benzylic and allylic. All the exact same type of analysis as Sn1.
Does the base affect the rate? What kind of bases will be preferred for E1? No, not in RDS typically weak and used as a solvent
Best solvent? protic
Sn1 and E1 occur together!! They will be considered equally major.
![<p>Starts with a molecule undergoing a reaction.</p><p>Elimination uses a base to attack an acidic hydrogen and then uses that to create a double bond between the two carbons. </p><p></p><p><u>Kinetics of the E1</u></p><p>What is the RDS & rate law? Step 1 (from carbonation) rate = k[Rx] </p><p> What kind of alkyl halides will be best for E1? Tertiary and secondary alkyl halides, as well as benzylic and allylic. All the exact same type of analysis as Sn1. </p><p> Does the base affect the rate? What kind of bases will be preferred for E1? No, not in RDS typically weak and used as a solvent</p><p> Best solvent? protic</p><p><strong>Sn1 and E1 occur together!! They will be considered equally major. </strong></p><p></p>](https://knowt-user-attachments.s3.amazonaws.com/5c211b28-e196-4e62-9308-fc66de1c21e6.png)
Selectivity Concerns in E1 and Zaitsev’s Rule
Rearrangements
Any time you form a carbocation.. Think about rearrangements.
→ C+ that is most stable (major products) other options HAVE to be favorable to occur.
Deprotonating at the adjacent position (which hydrogen do you pull and which double bond do you make).
Zaitsev’s Rule
The most stable alkene product will be major product
In E1 elimination reactions the most stable alkene usually predominates
most stable C+ atom → kinetics
Most stable alkene (conjugation and substitution)
Tip: when looking at shifts, look at atom connectivity!!

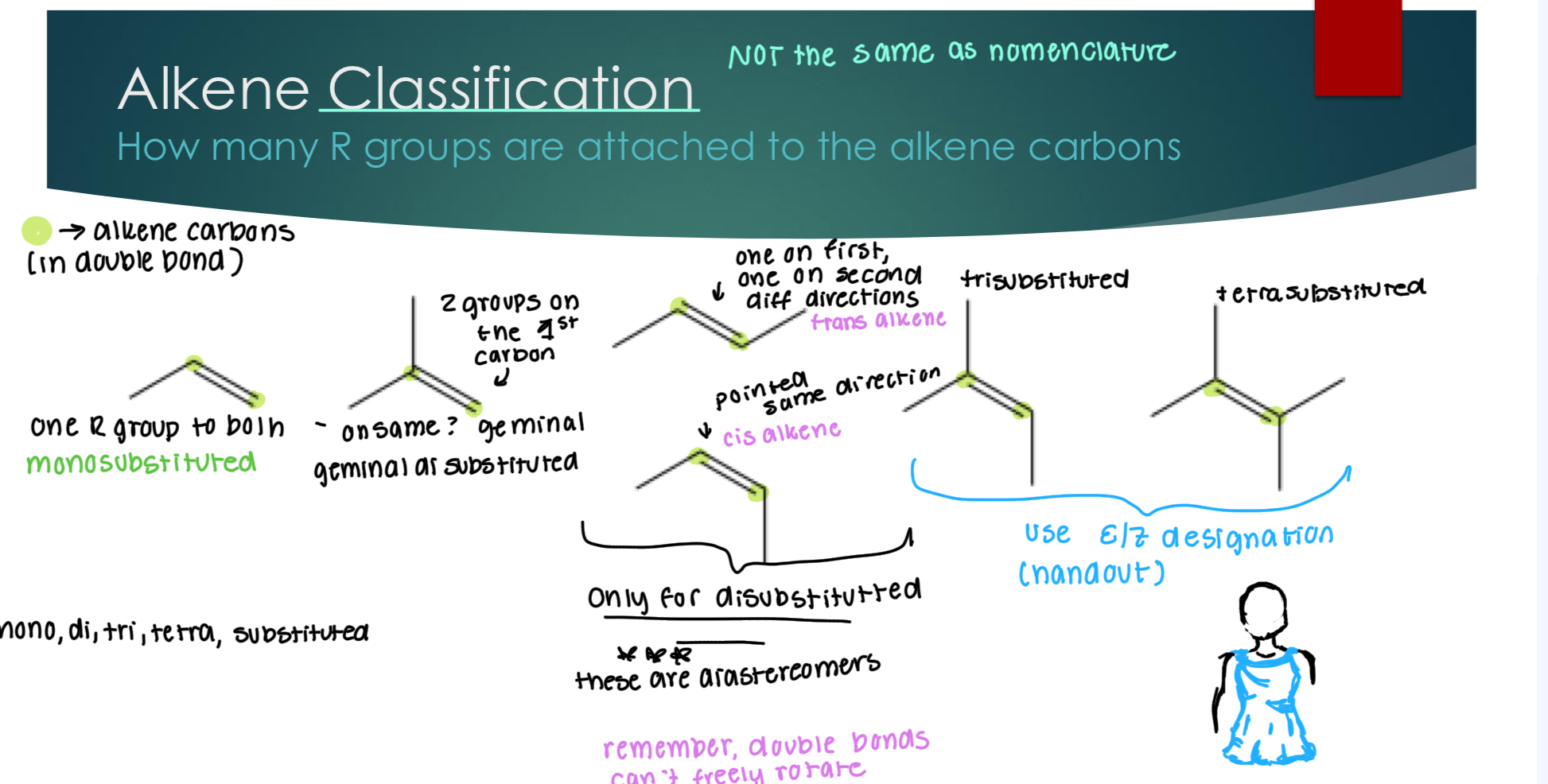
Alkene Classification from slides: How many R groups are attached to the alkene carbons
Look at handout, but basically the disubstituted substitutions are diastereomers. Geminal is when the two groups are off a singular carbon. Basically, look at how many groups are attached to the double bonded carbon.
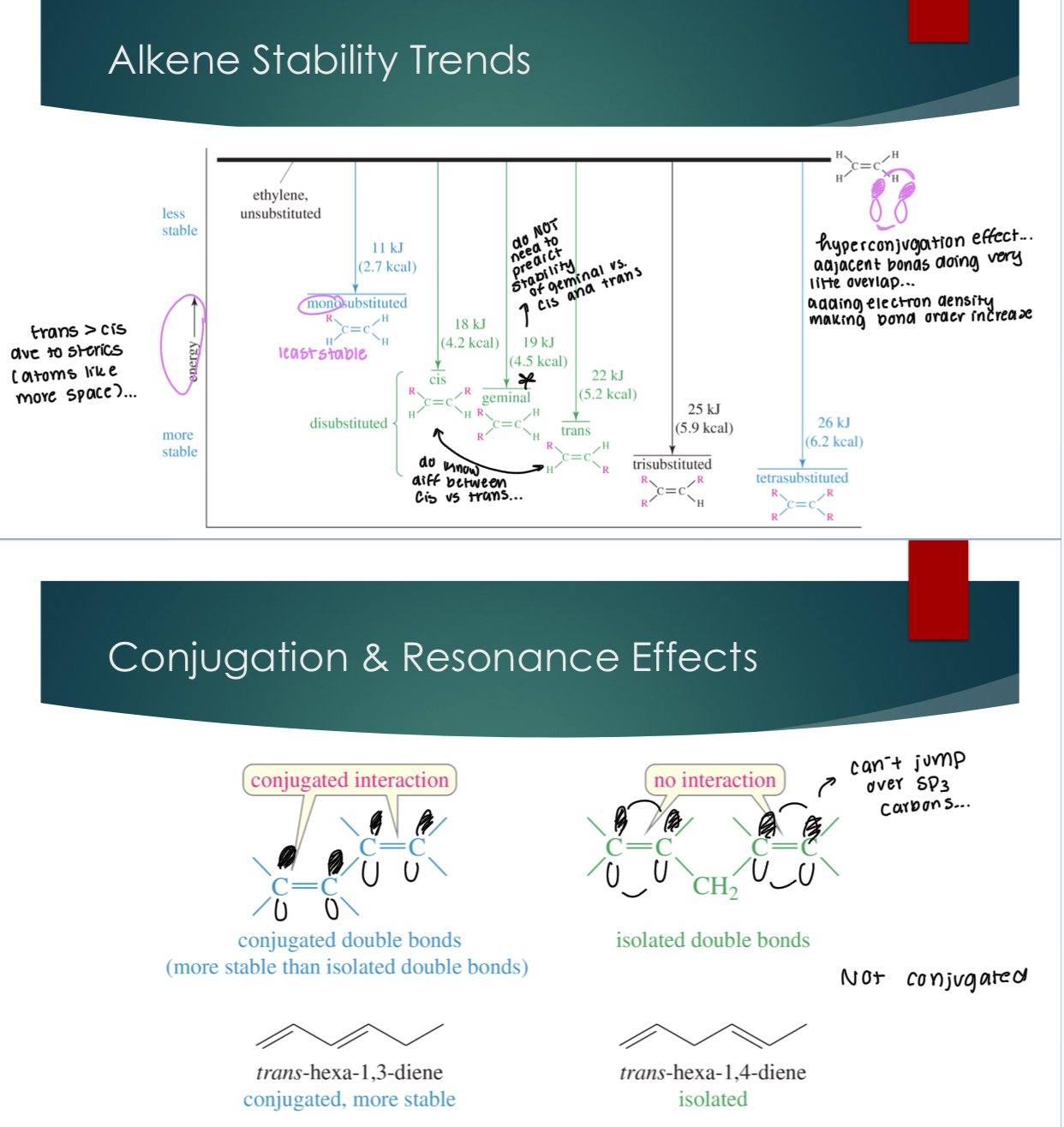
Alkene stability, conjugation, and resonance effects
Know that mono is the most unstabled and tetra is the most stable. In terms of cis and trans, trans is more stable, because of steric (atoms like space).
Why is tetra more stable: hyperconjugation effects… Adjacent bonds doing very little overlap. Adding electron density, making bond order increase.
Conjugated interactions mean that when there are more double bonds adjacent to the made double bonds from the E1 mechanism.
E2 Elimination Mechanism + Kinetics
What are the similarities to Sn2? What are the differences?
it’s one step: (concerted)
but, it’s not pentavalent since it only has 4 groups due to the double bond. NOTE: don’t count the double bonds as two groups.
What is the RDS & rate law? k[B][Rx]
What kind of alkyl halides will be best for E2?
No pentavalent, C 3° is okay for this reaction. Alignment is important, though.
Does the base affect the rate? What kind of bases will be preferred for E2?
Yes, want a strong base/ nucleophile small or bulky will work..
Best solvent?: aprotic
Use it to determine what is slightly faster or slower.
![<p>What are the similarities to Sn2? What are the differences?</p><p>it’s one step: (concerted) </p><p>but, it’s not pentavalent since it only has 4 groups due to the double bond.<strong> NOTE: don’t count the double bonds as two groups. </strong></p><p></p><p>What is the RDS & rate law? k[B][Rx] </p><p> What kind of alkyl halides will be best for E2? </p><p>No pentavalent, C 3° is okay for this reaction. Alignment is important, though. </p><p> Does the base affect the rate? What kind of bases will be preferred for E2? </p><p>Yes, want a strong base/ nucleophile small or bulky will work.. </p><p> Best solvent?: aprotic</p><p>Use it to determine what is slightly faster or slower.</p>](https://knowt-user-attachments.s3.amazonaws.com/a61d28ca-e4d3-4503-94a5-7b124440bc01.png)
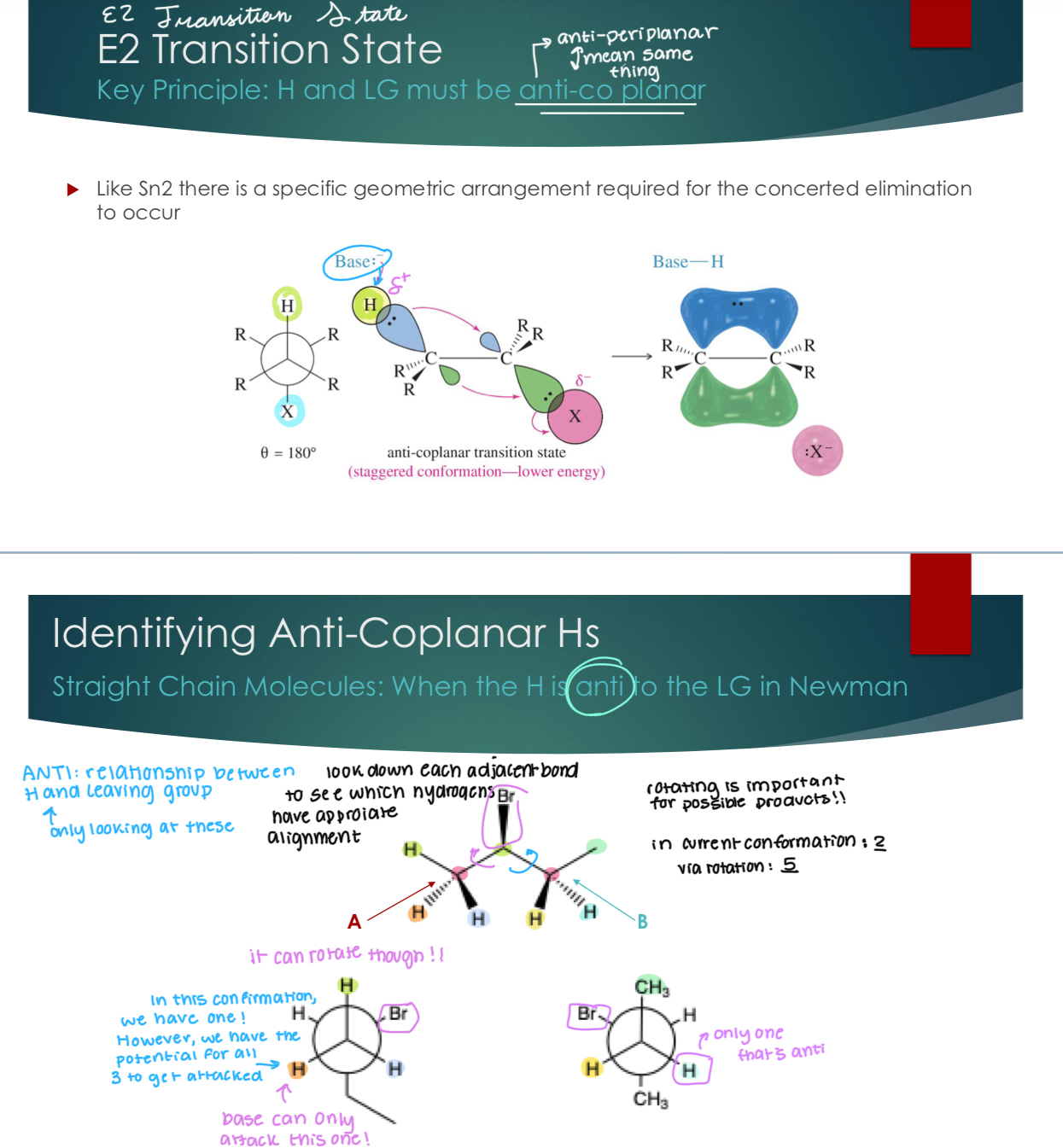
E2 Transition State
Key Principle: H and LG must be anti-co planar (anti-peri planar) and identifying them
Like Sn2 there is a specific geometric arrangement required for the concerted elimination to occur
Straight Chain Molecules: When the H is anti to the LG in Newman
ANTI: only looking at relationship between H and LG.
Rotating is important for possible products. In current conformation there are 3 possible outcomes, via rotation there are 5.
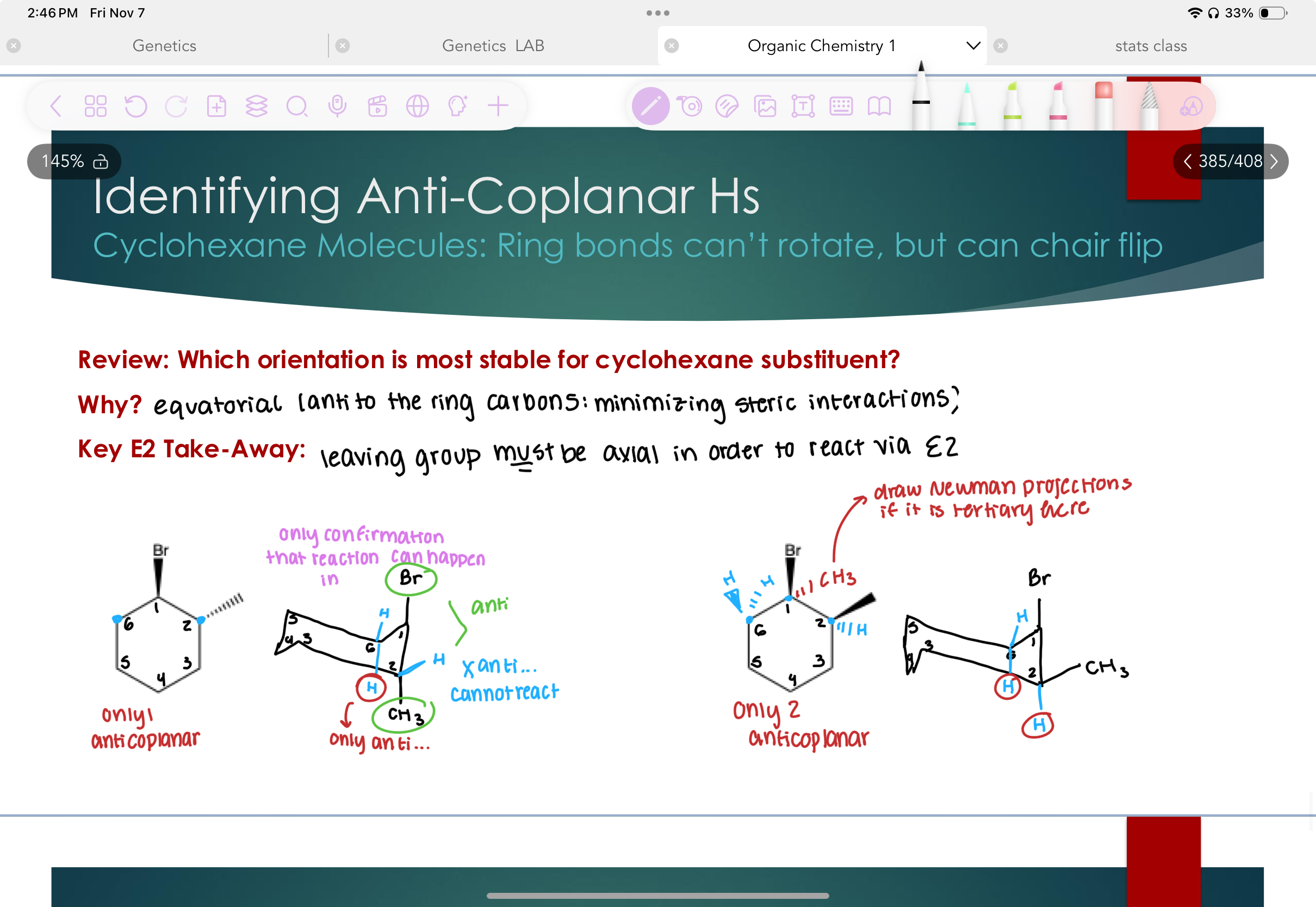
Indentifying Anti-Coplanar H’s in cyclohexanes.
Look at chair flip!!
Eq is more stable, because there is more space and it minimizes steric interactions.
LG MUST be axial in order to react via E2.
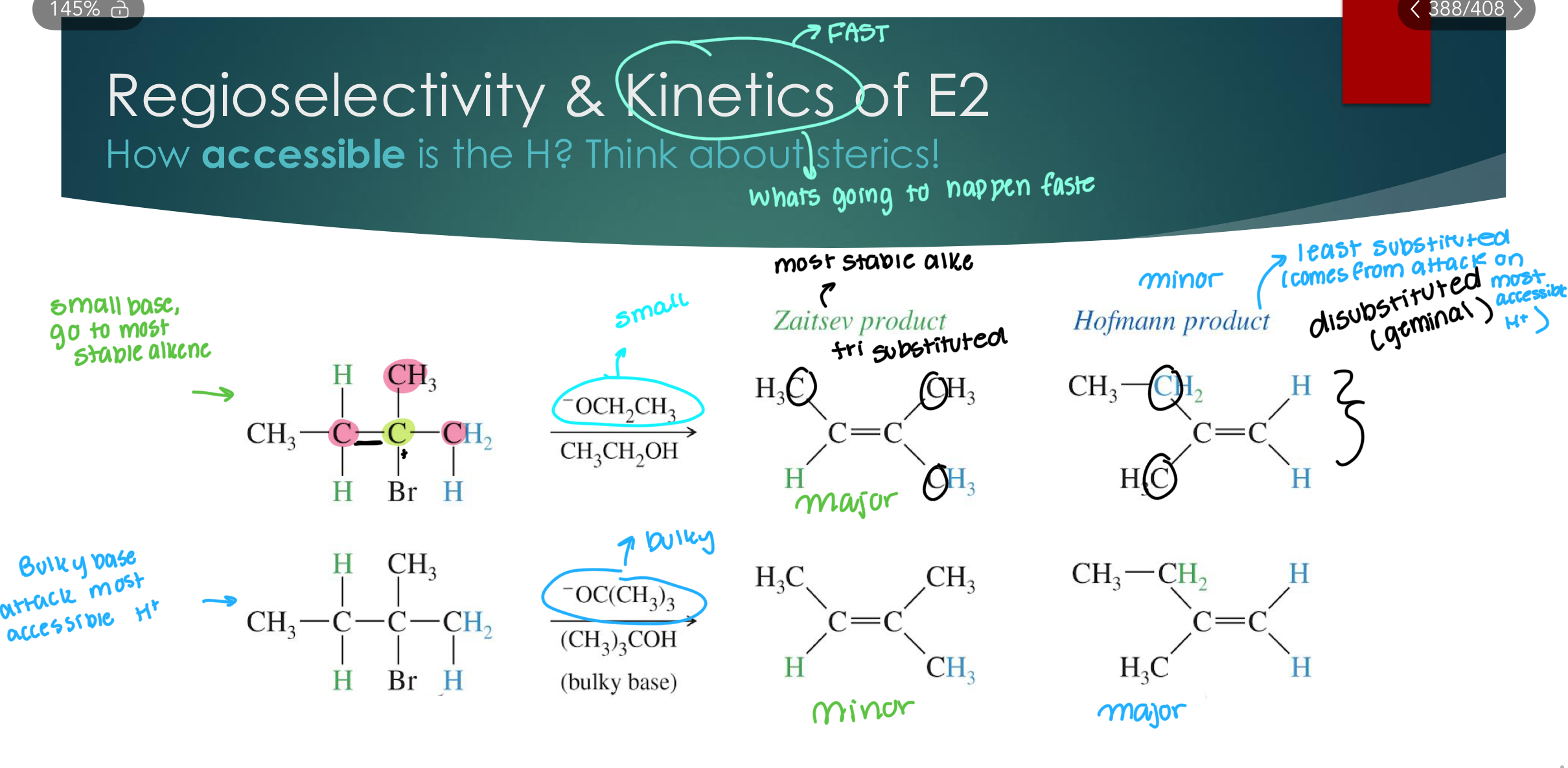
Regioselectivity
Recall: A reaction is regioselective if bond formation is preferred at one atom over other possible
atoms
SN2: Substitution only occurs at C attached to the LG: NOT regioselective (only one possibility)
Sn1: Substitution can occur wherever there is a C+ : Yes! When rearrangement occurs: regioselective for most stable C+.
E1: Elimination can occur at any acidic (adjacent) H to a C+: Yes! When rearrangement occurs: regioselective for most stable C+. 2nd is choosing H+ that makes the most stable alkene.
E2: Elimination can occur at any H anti-coplanar to the LG: Yes! when there are anti-coplanar hydrogens…
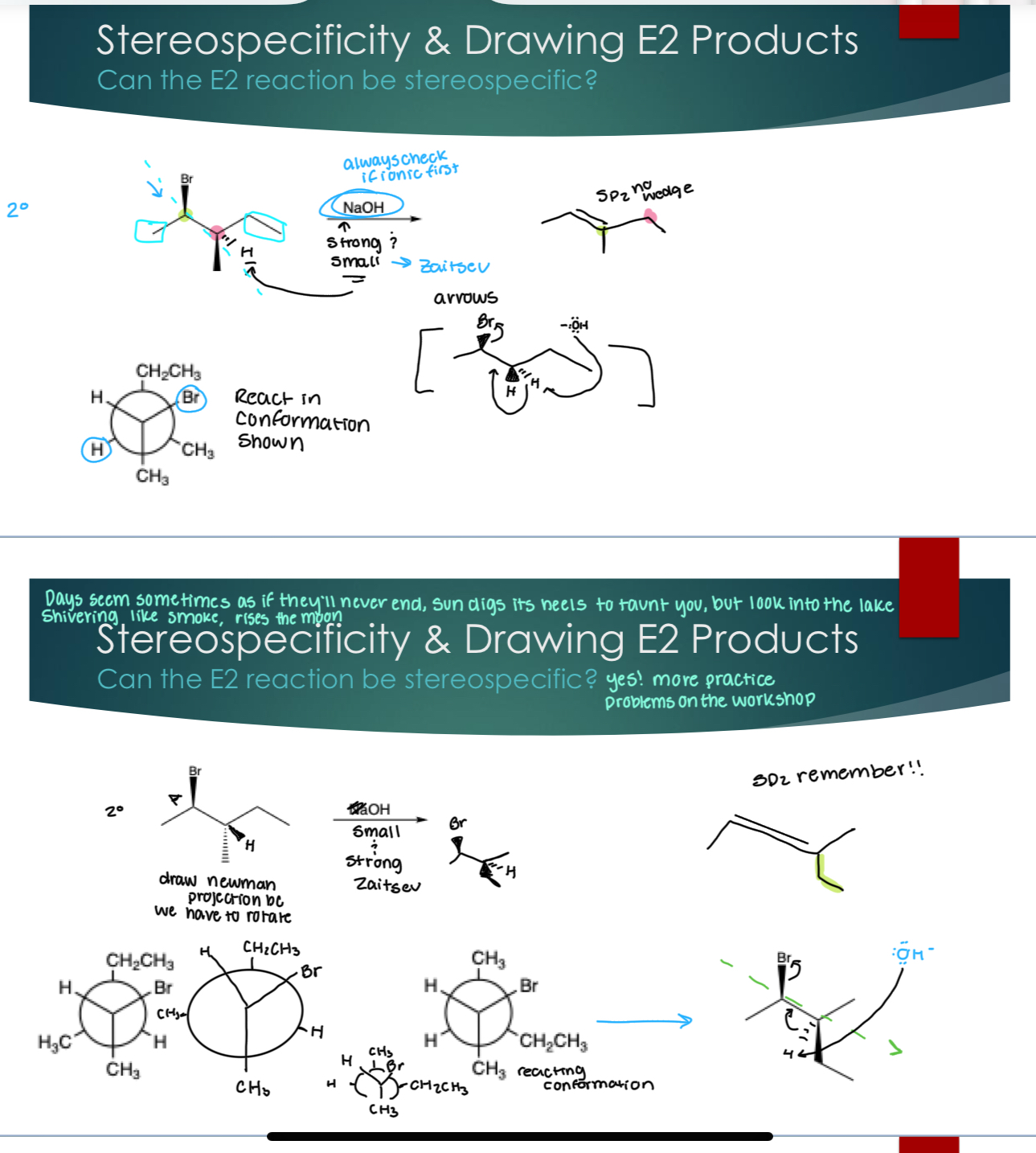
Stereospecificity
Recall: A reaction is stereospecific if different stereoisomers lead to different products
Sn2: Yes! It has inversion of stereocenter
Sn1: C+ removes stereochemistry (og never matters) NOT stereospecific
E1: C+ removes stereochemistry NOT stereospecific
E2: Yes!! anti-coplanar shit - see picture
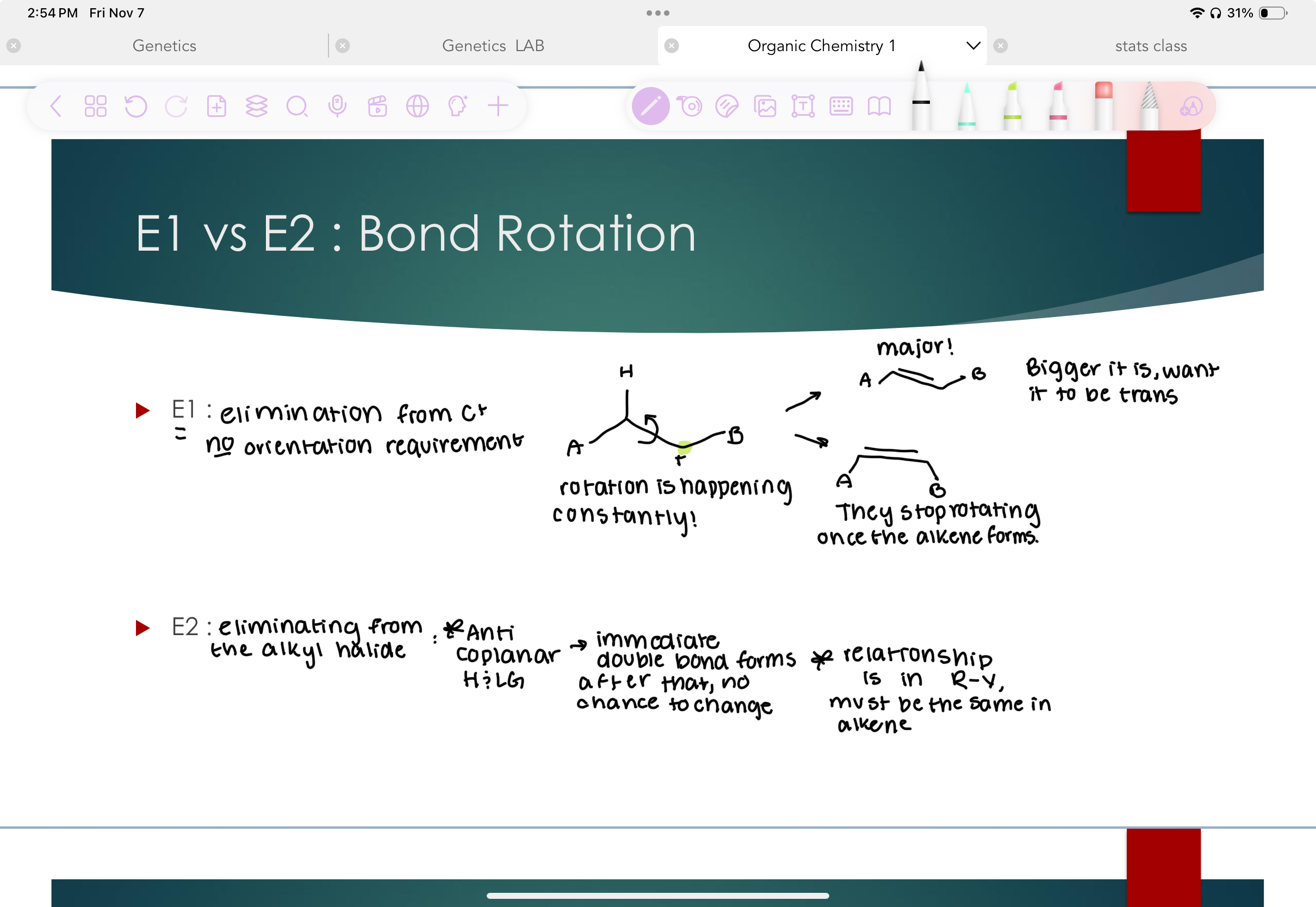
E1 vs. E2 Bond rotation
E1: elimination from C+ no orientation requirement, rotation is constantly happening until the alkene forms, bigger = trans
E2: elimination from the alkyl halide ** anticoplanar H-LG → immediate double bond forms after that, no change relationship is in R-X must be the same in alkene
Summary table for mechanisms and nucleophiles
Summary of major v minor products
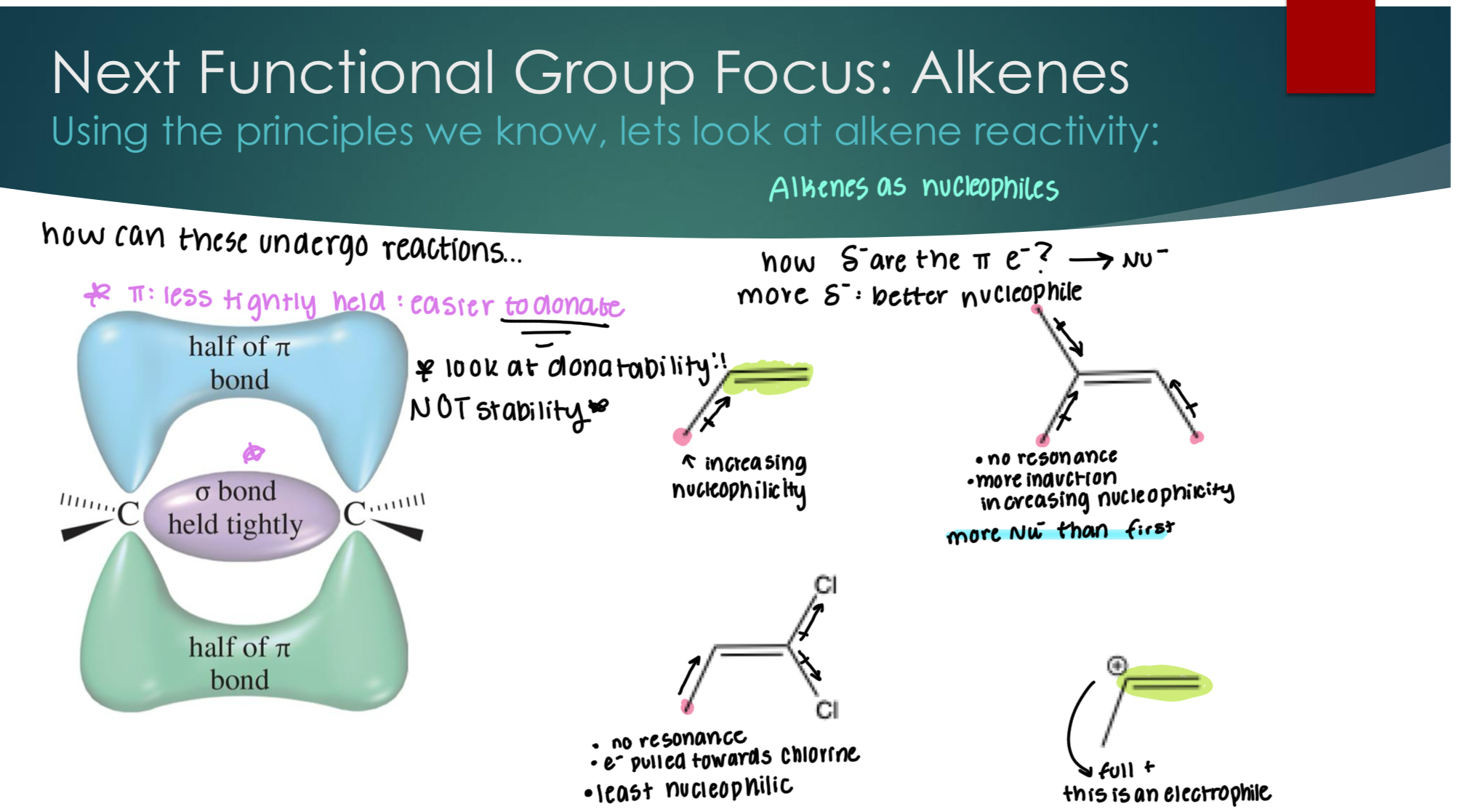
Next Functional Group Focus: Alkenes Using the principles we know, let’s look at alkene reactivity:
Pi bonds have electrons that are easier to donate. AKA LOOK AT DONATABILITY.
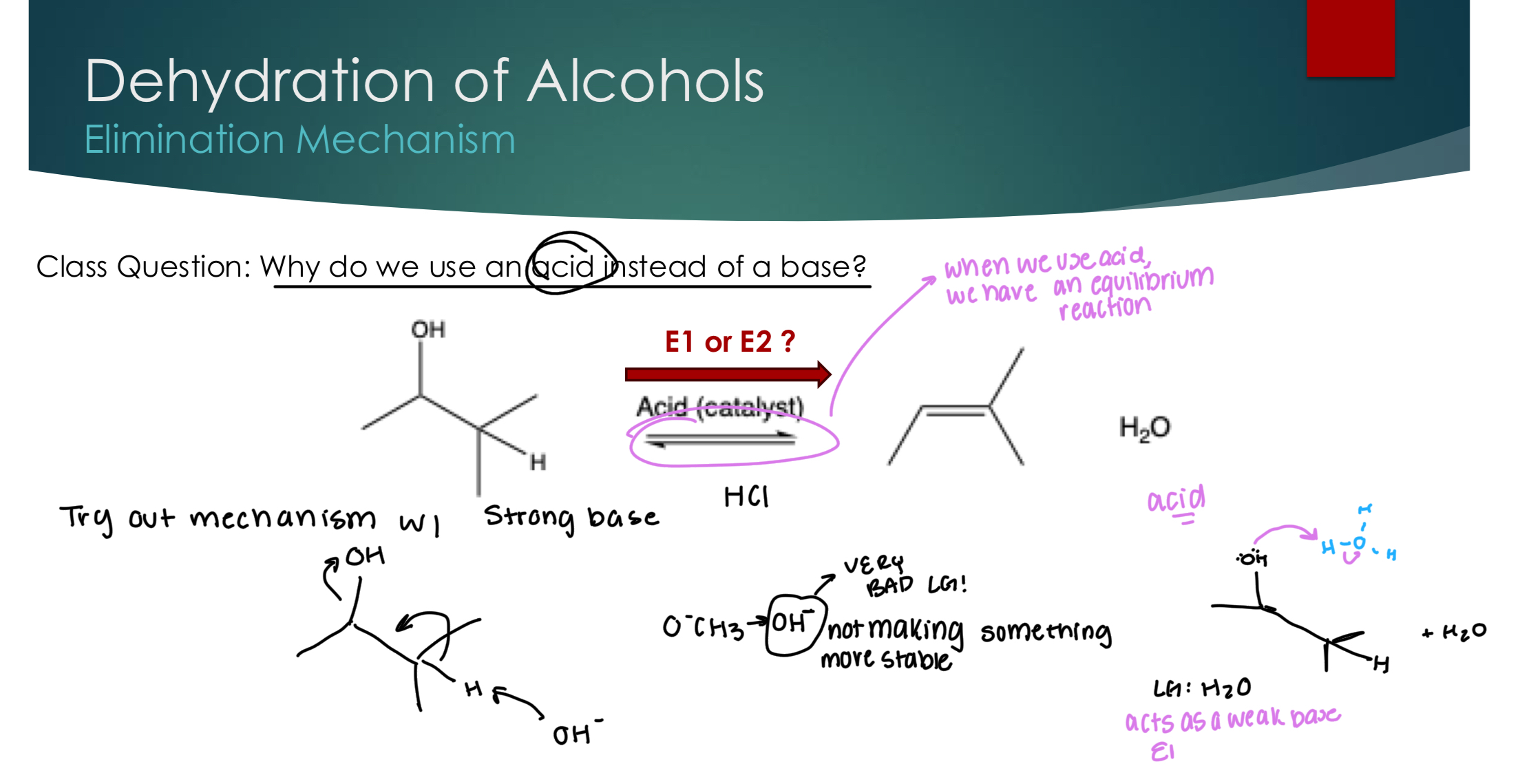
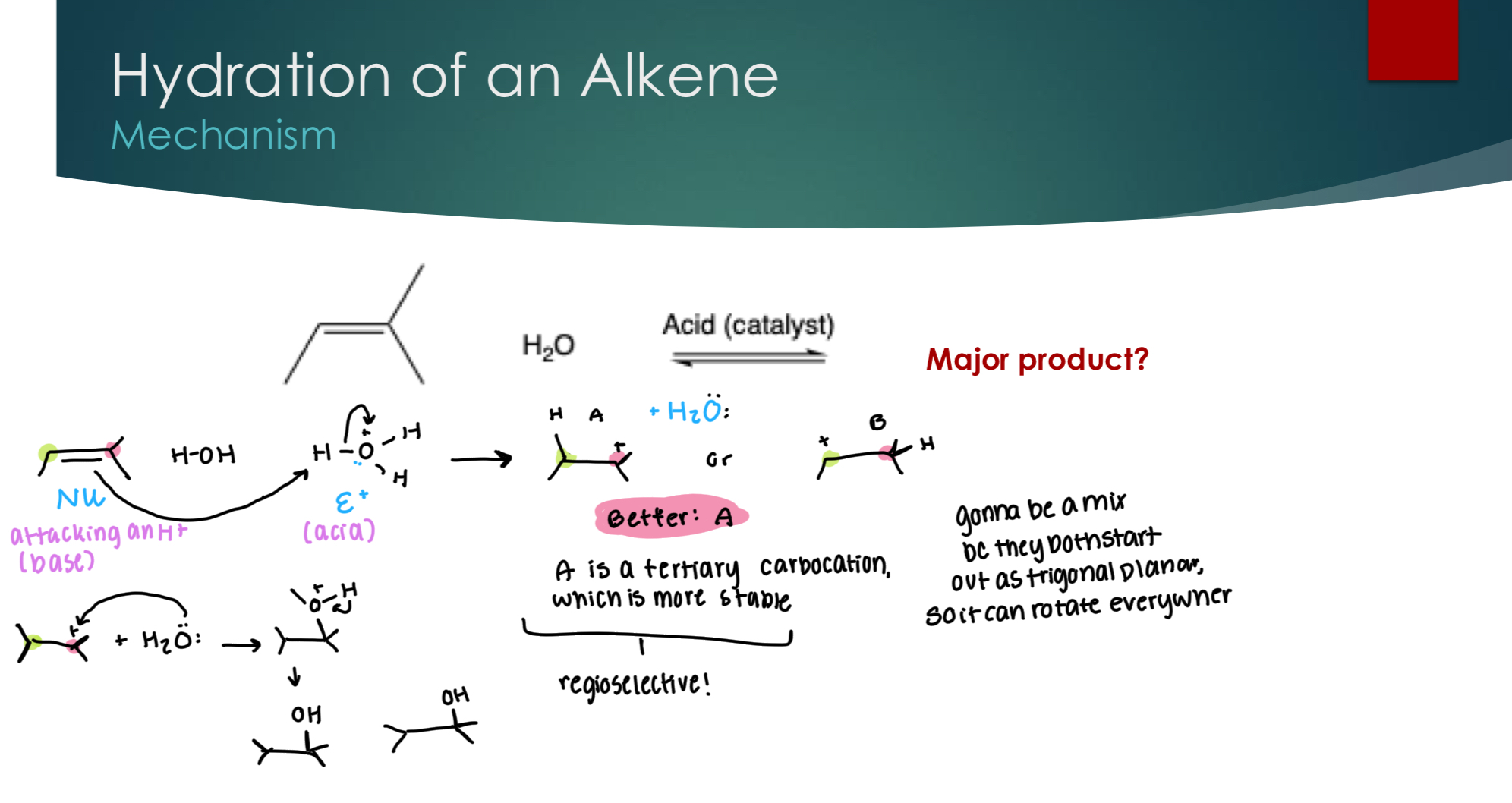
Dehydration of Alcohols: elimination mechanism
We use an acid when we have an equilibrium reaction, because OH is a terrible LG, and so we need H2O to act as a weak base to attack a H+ ion to make a double bond and kick off the OH.
LeChatlier’s Principle: wants to undo any change that you do..
If you want to favor the alcohol: add lots of H2O
If you want to favor making the alkene, heat it up! (remove H2O)
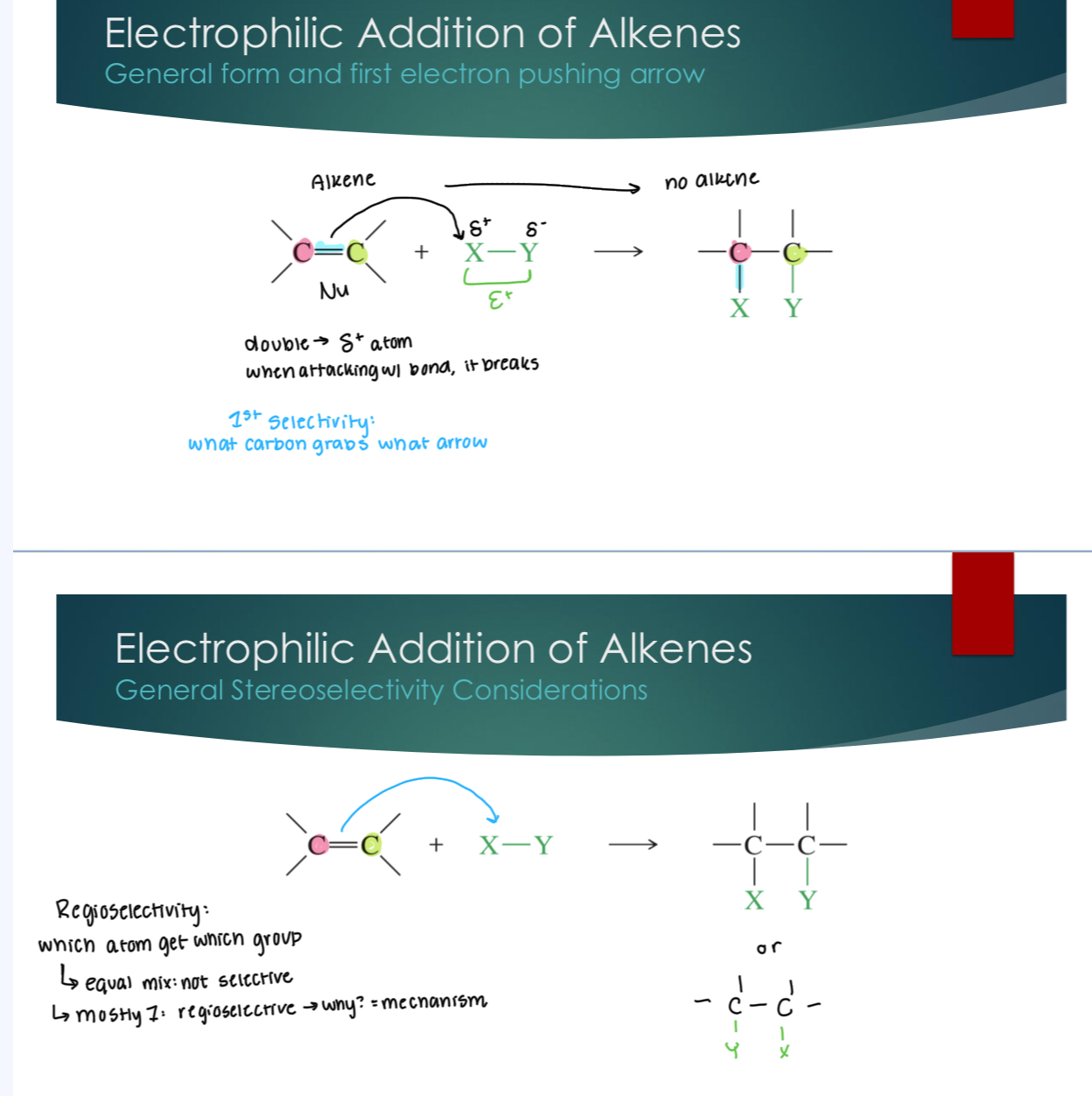
Electrophilic Addition of Alkenes
Double → Partial + atom, when attacking w/ bond, it breaks. 1st point of selectivity: what carbon grabs what arrow.
Regioselectivity: which atom gets which group
equal mix (not selective)
Mostly one over the other (regioselective → why? = mechanism)
Stereoslectivity: relationship of "X” and “Y” e.g.
if X and Y are on the same side (bond type) SYN
X and Y are on opposite sides: ANTI
Can be stereocenters w/o double bond because SP3, but not concerned with exact stereocenter configs.
Hydration of an Alkene
Forming a carbocation, going to be based off of the most stable C+. In this example, A is a tertiary carbocation, which is more stable. Making it regioselective.
Going to be a mix between wedge and dash because it starts out as trigonal planer.
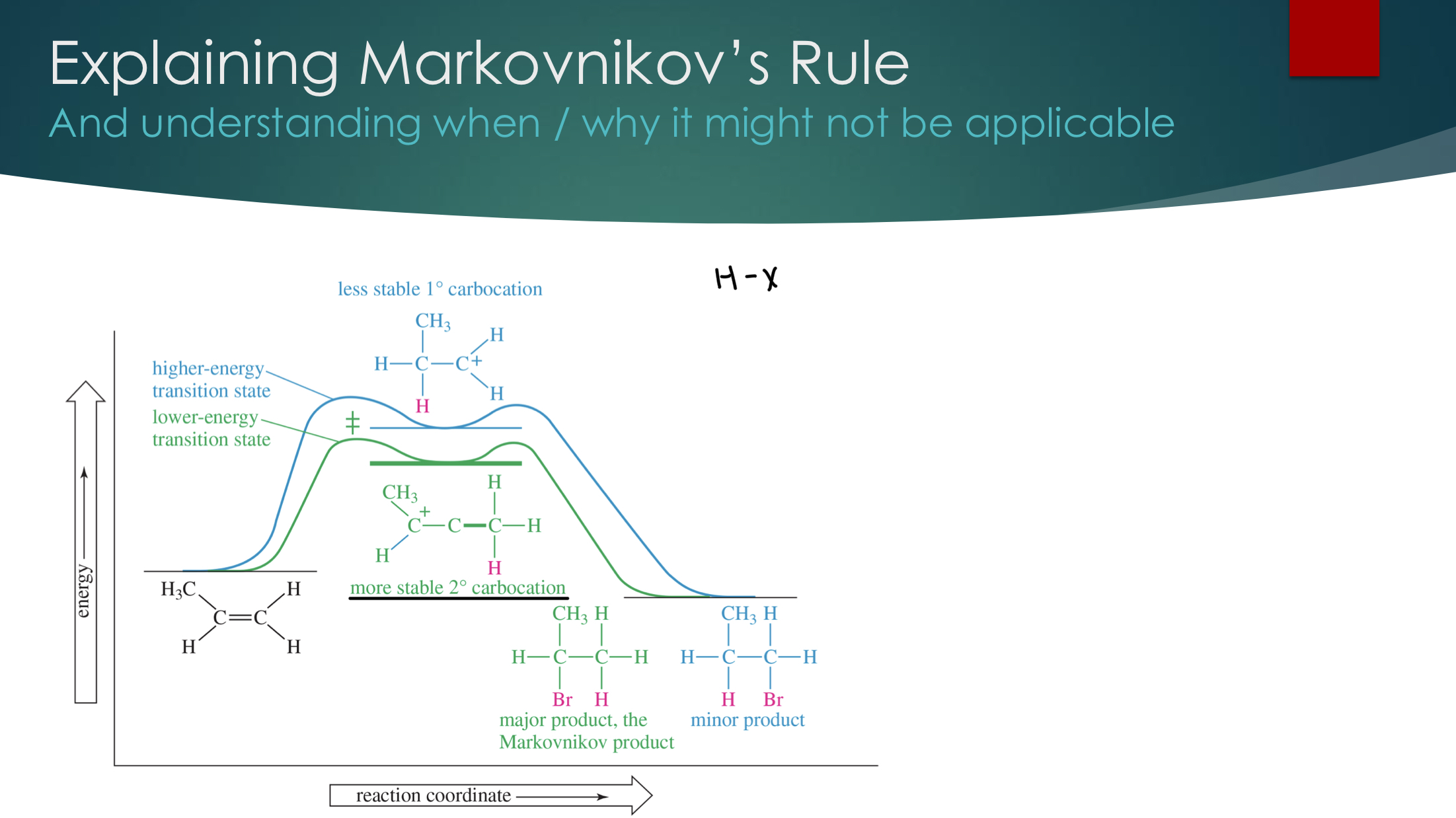
Determining Regioselectivity: Markovnikov’s Rule
In the addition of an acidic proton to an alkene, the proton will add to the alkene carbon that is bonded to the most H atoms
Another way to phrase it… when we have H-Y, H on the least substituted alkene C and “Y” on the most substituted alkene carbon.
We can justify this using Hammond’s postulate where a more stable carbocation lowers the activation energy and makes it faster.
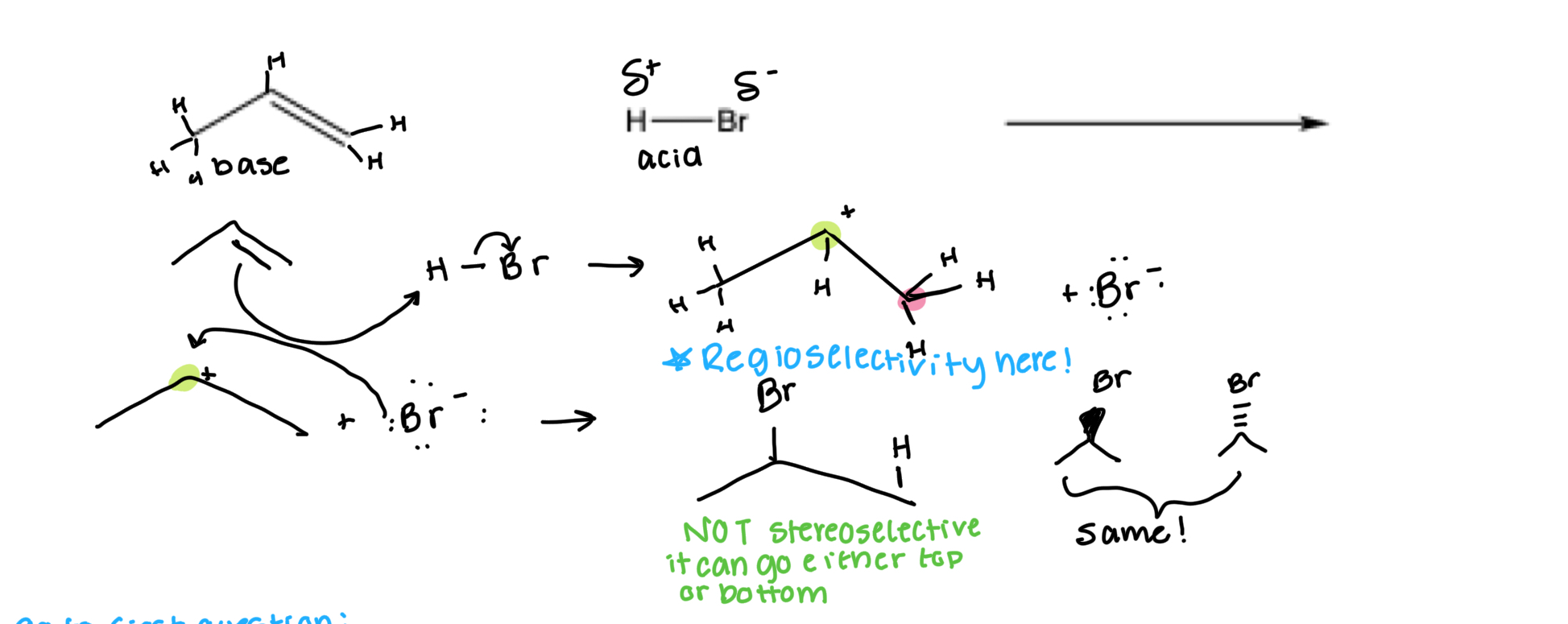
Hydrohalogenation
Stereoselectivity: We got a mix… NOT selective: adding to a flat C+
Regioselectivity: Marks Rule! X on more substituted C… so yes!
Without drawing the mechanism, draw the major product(s) of the following reaction:
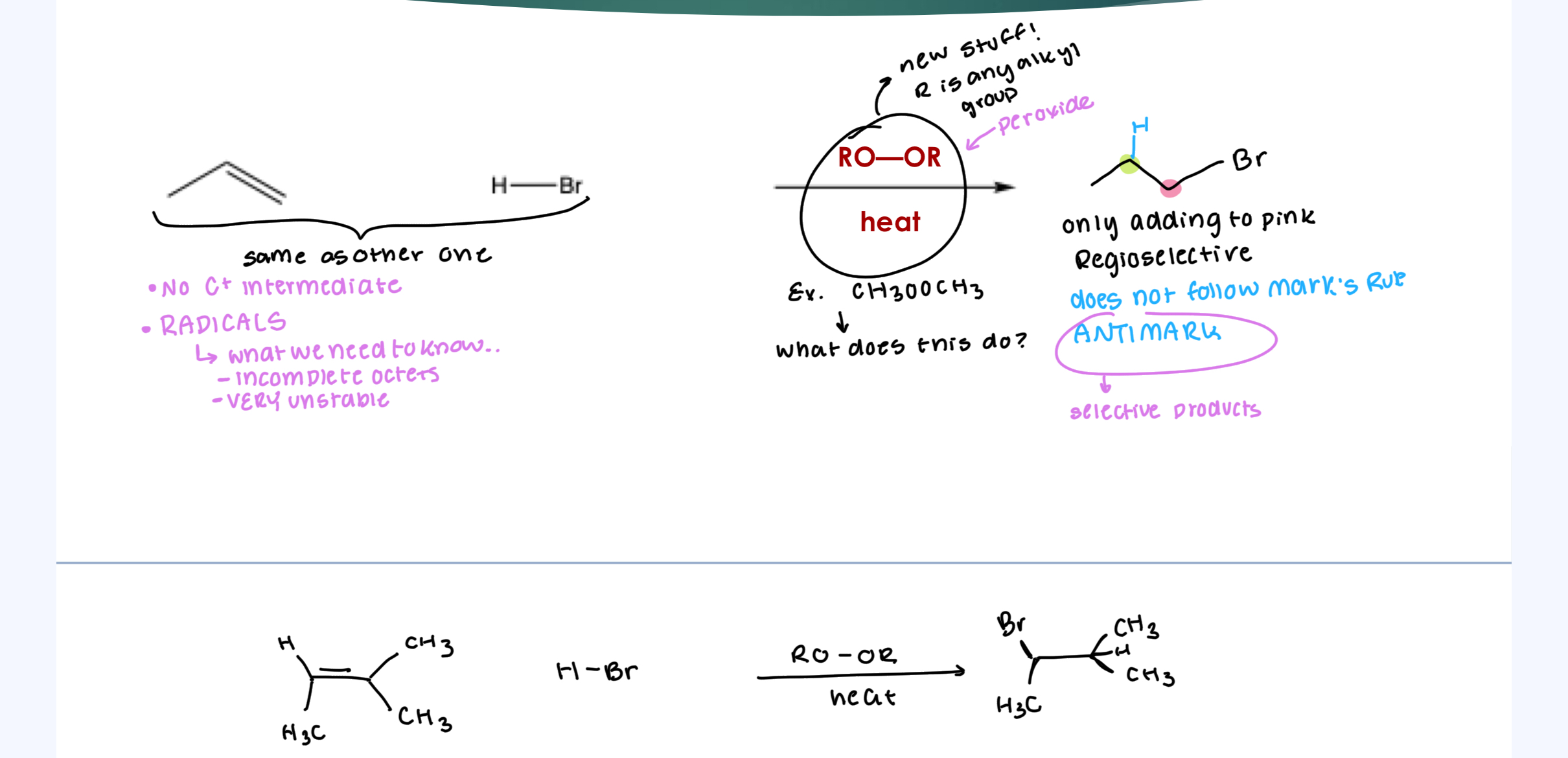
The Radical Hydrohalogenation Reaction
No C+ intermediate
RADICALS
What we need to know about these:
Incomplete Octets
VERY unstable
Stereoselectivity: Like C+: no stereoselectivity… doesn’t care about top or bottom.
Regioselectivity: ANTI MARK (most stable Radical intermediate
Need to recognize the EtOOEt + heat for the radical
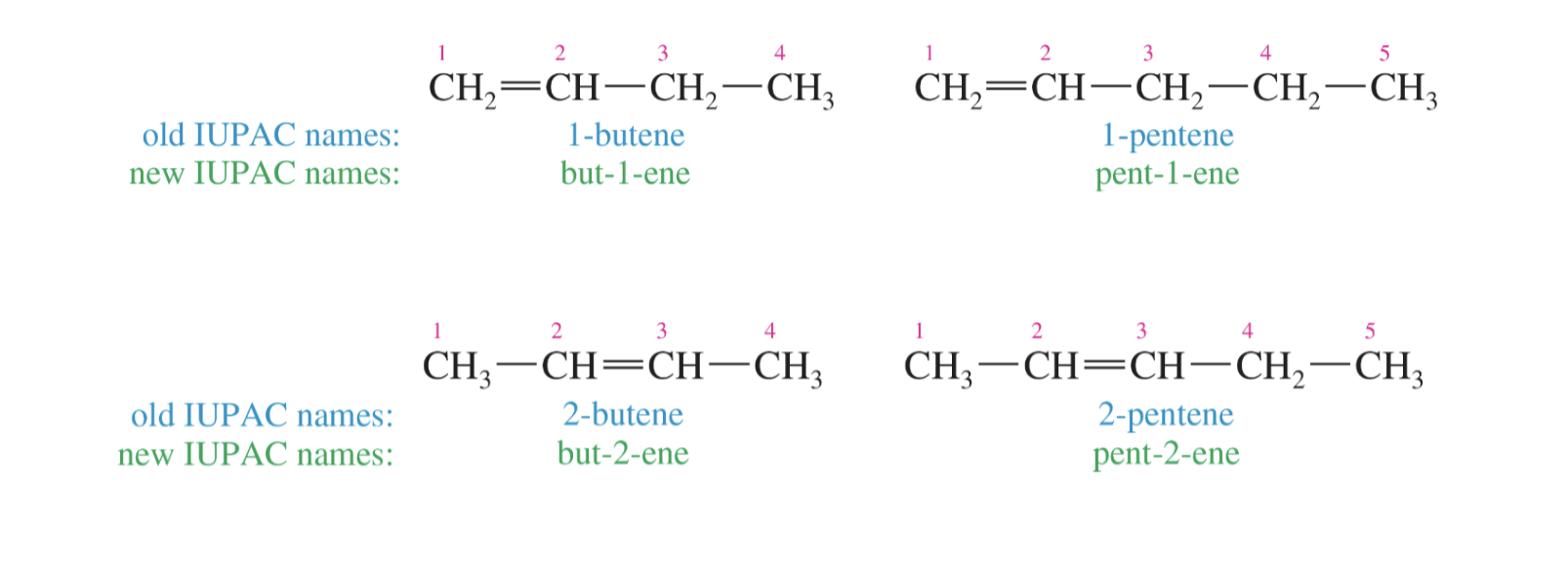
Naming Alkenes: basics
1. Base name is based on the longest carbon chain and the suNix for the base name corresponds to the highest priority functional group present.
**New: the longest carbon chain must include the alkene
**New: alkenes are higher priority than alkanes, so suffix is ene
Carbon number prefix for longest carbon chain + C# of first alkene C + ene
2. Alkyl and halogen substituents are named the same, with the same best practices.
**With an alkene present, the best practice is prioritization for numbering the alkene (over other substituents) with the lowest carbon numbers possible
Naming Alkenes: steps and rules
Steps and Rules:
1. Find the longest carbon chain, containing the alkene and number the chain so that the alkene carbons have the lowest numbers possible.
2. Write the base name:
New (use this one): carbon number prefix - number of the first alkene carbon - ene Old (just so you are aware): number of the first alkene carbon – carbon number prefix + ene
3. If substituents are present, add the substituent names with their carbon number to the front of the base name in alphabetical order.
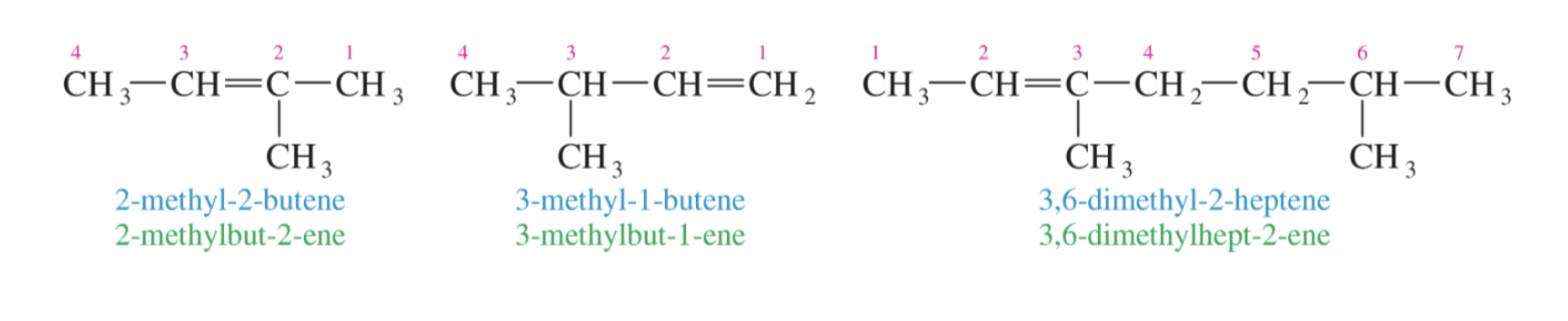
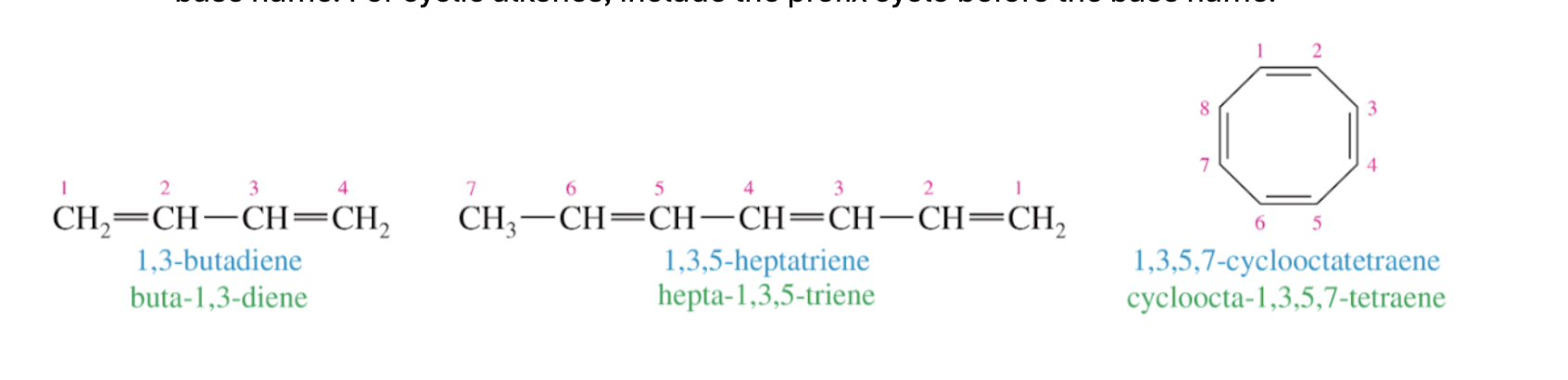
4. If multiple alkenes are present, like how we treat multiple of the same substituent, include the carbon numbers for both alkenes and use the prefix “di” or “tri” etc. in the base name. For cyclic alkenes, include the prefix cyclo before the base name.
Stereochemistry of Alkenes
Due to the restricted rotation of double bonds, some alkenes have the possibility of having geometric isomers, a type of diastereomer.
Cis/Trans: Simple alkenes (only one R group per alkene carbon) that contain a hydrogen atom directly attached to each alkene carbon are often classified using cis/trans. However, this designation becomes less clear when multiple carbon groups are attached to the same alkene carbon.
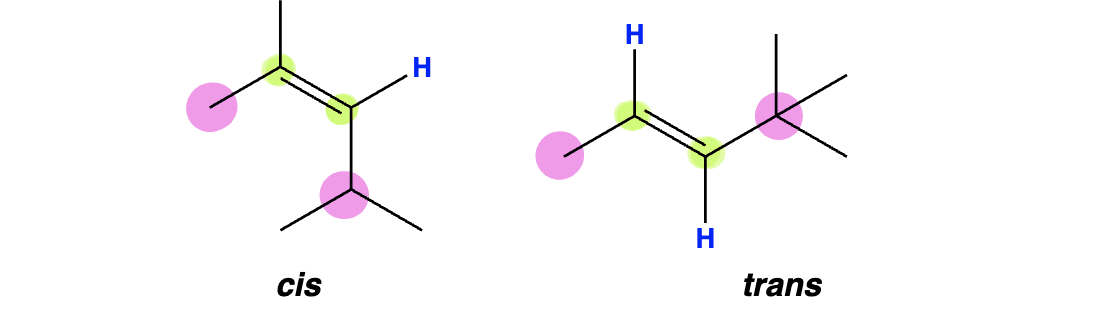
E/Z Designation: Alkenes containing multiple (and unique) carbon groups attached to the same alkene carbon are classified with a more standardized system based on the same IUPAC prioritization rules that we used in classifying stereocenters as R or S.
Step 1: Rank the groups attached to the same alkene carbon in priority (1 = higher priority and 2 = lower priority) using the exact same IUPAC priority rules that we used to determine whether a stereocenter was R or S
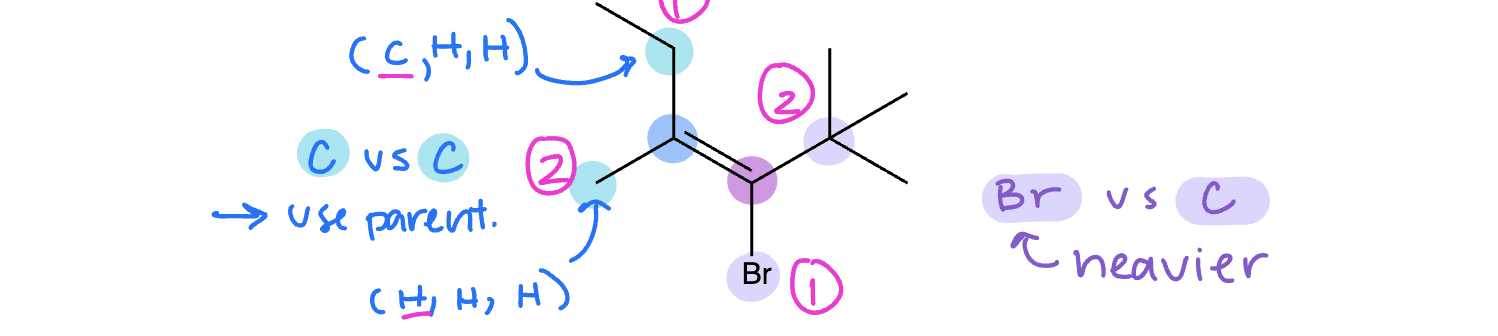
Step 2: Look at the relationship between the number one priority groups on each alkene.
If the number 1 groups are on the same side of the alkene (cis type relationship) then the alkene is designated as Z. (Potentially helpful mnemonic: they are on zee same side)
If the number 1 groups are on opposite sides of the alkene (trans type relationship) then the alkene is designated as E
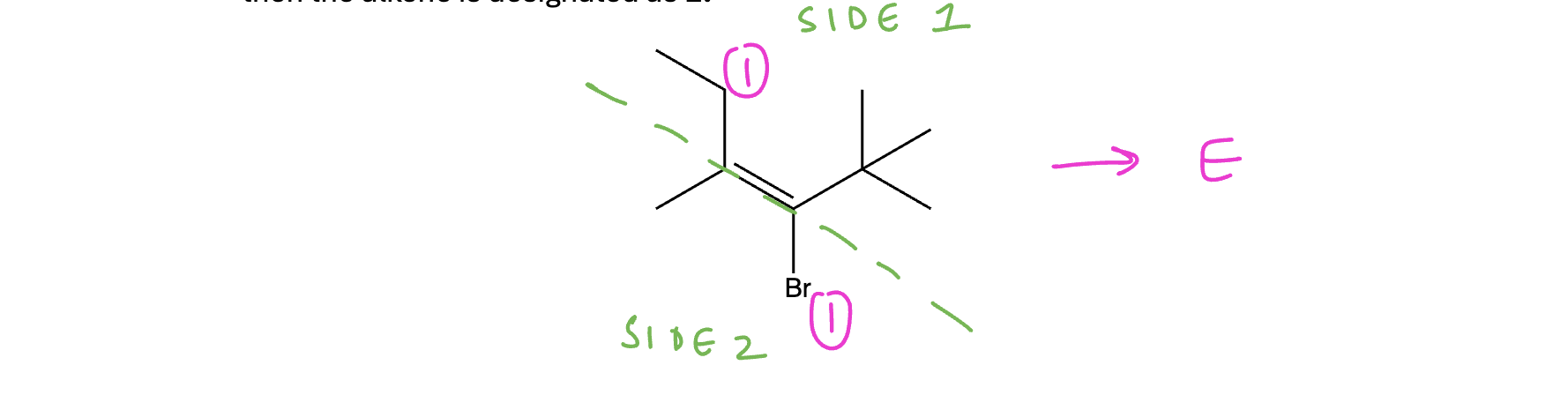
Example naming an alkene with E/Z designations
If the alkene can be classified as either cis/trans or E/Z, then this designation is included in parentheses at the front of the name
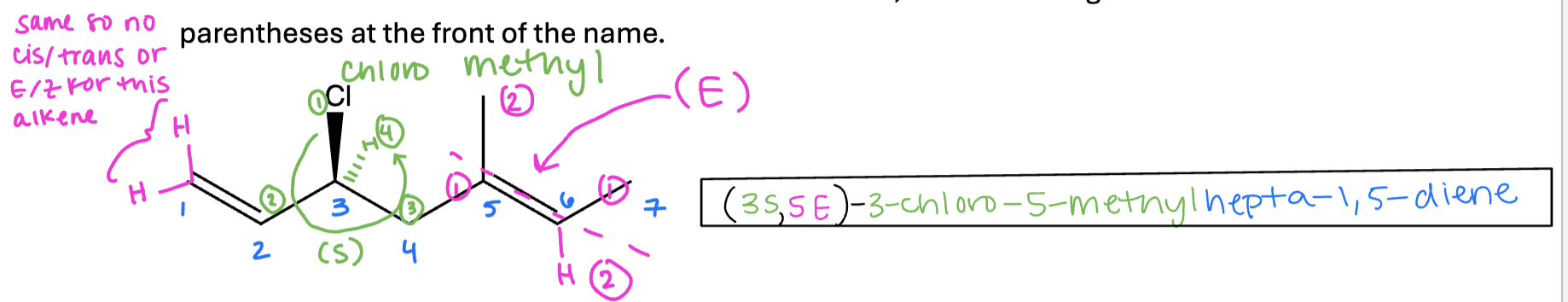
Alkenes in Substituents
In some cases with multiple alkenes, it is not possible to include both alkenes in one longest chain / base name. In this case, one alkene will be treated as substituent. When
alkenes are substituents, slightly different naming rules apply (like how we refer to a one carbon substituent as methyl instead of methane).
For this class, you only need to name two types of alkene substituents:
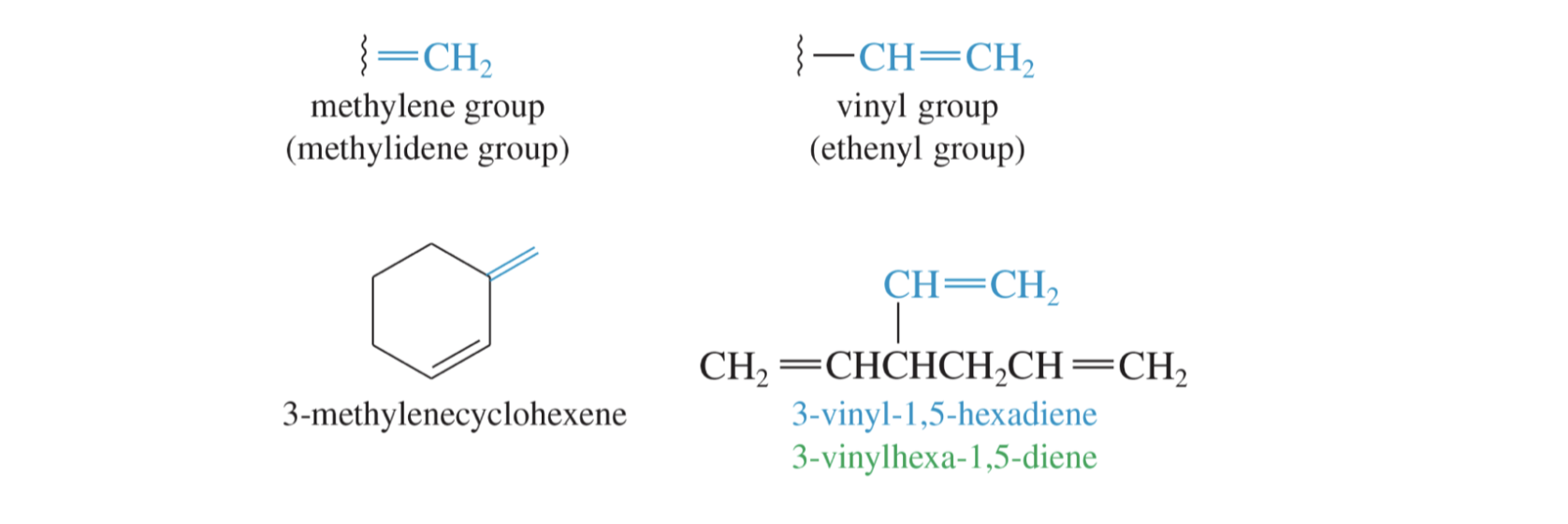
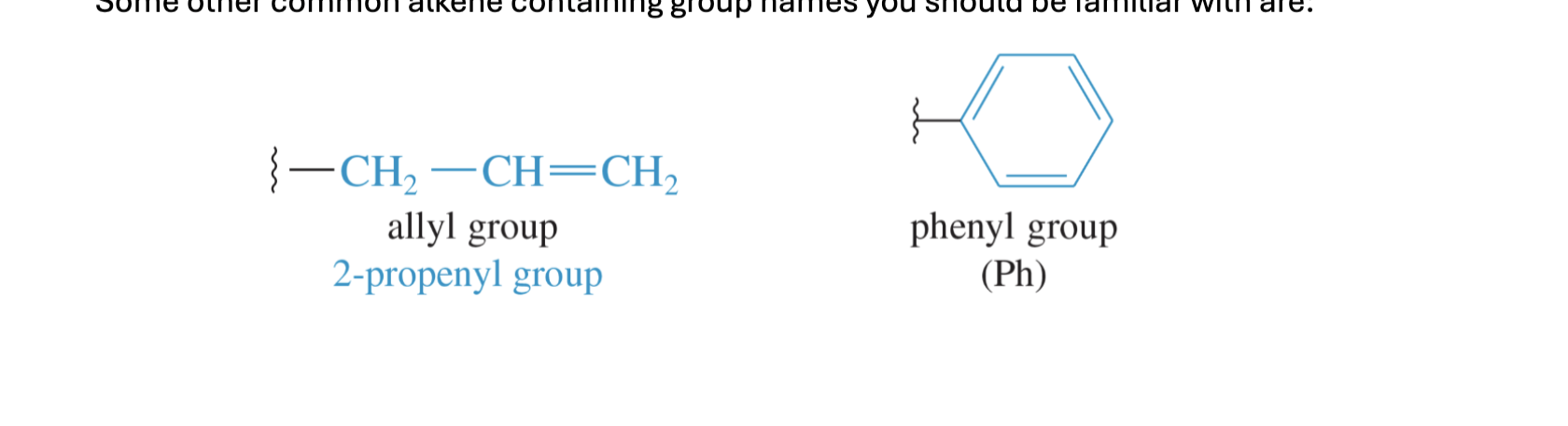
Some other common alkene containing group names you should be familiar with are:
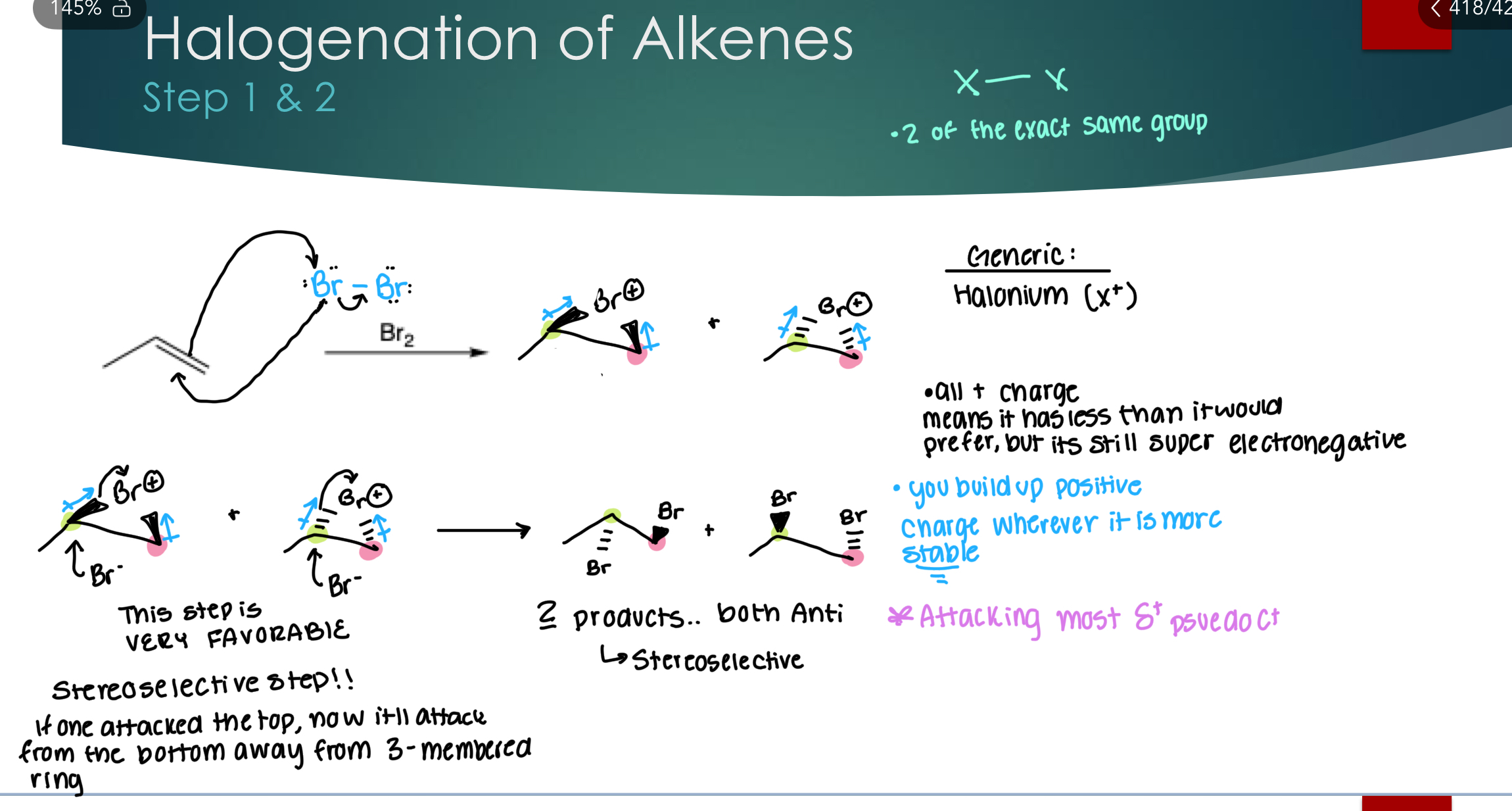
Halogenation of Alkenes
Stereoselectivity: Yes! Stereoselective for Anti because we are attacking Sp3 C, selective attack opposite the bulky 3-mem ring.
Regioselectivity: Yes! 1st bromine is always going to the same position, but no bc same group is added. 2nd bromine always attacks at the same stable C+
Without drawing the mechanism, draw the major product(s) of the following reaction:
*Note: Br is still electronegative with the + charge and still pulls electrons.
The second Br ALWAYS attacks the most stable carbocation. This is a stereoselective step!! If one attacked the top, now it’ll attack from the bottom, away from the 3-membered ring.
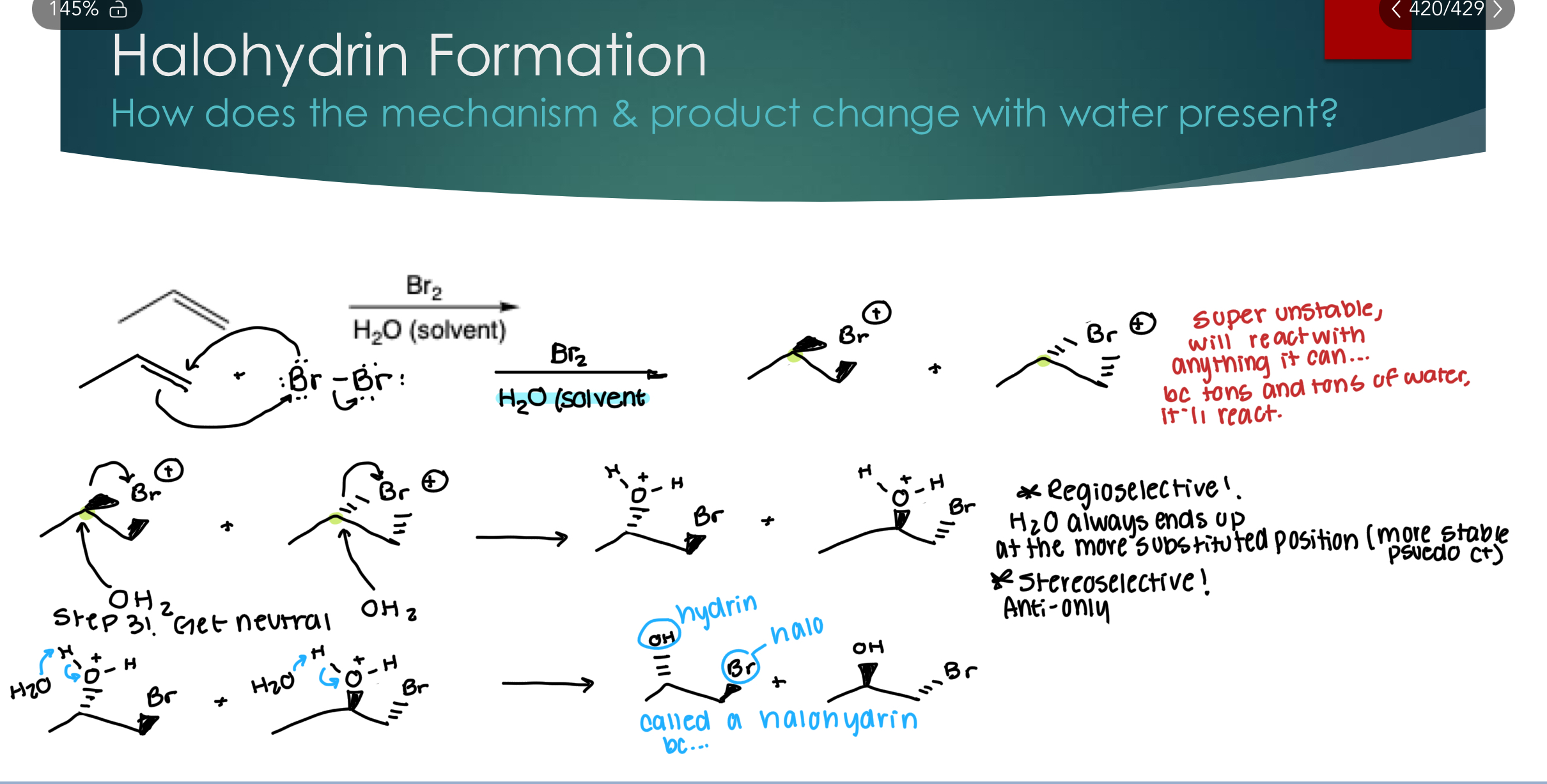
Halohydrin Formation
Stereoselectivity: Yes! ANTI: (same justification as halogenation)
Regioselectivity: Yes!! Water always ends up at the more substituted position (because its the more stable Pseudo C+) because it’s super unstable, will react with anything it can… bc tons and tons of water, it’ll react.
Without drawing the mechanism, draw the major product(s) of the following reaction:
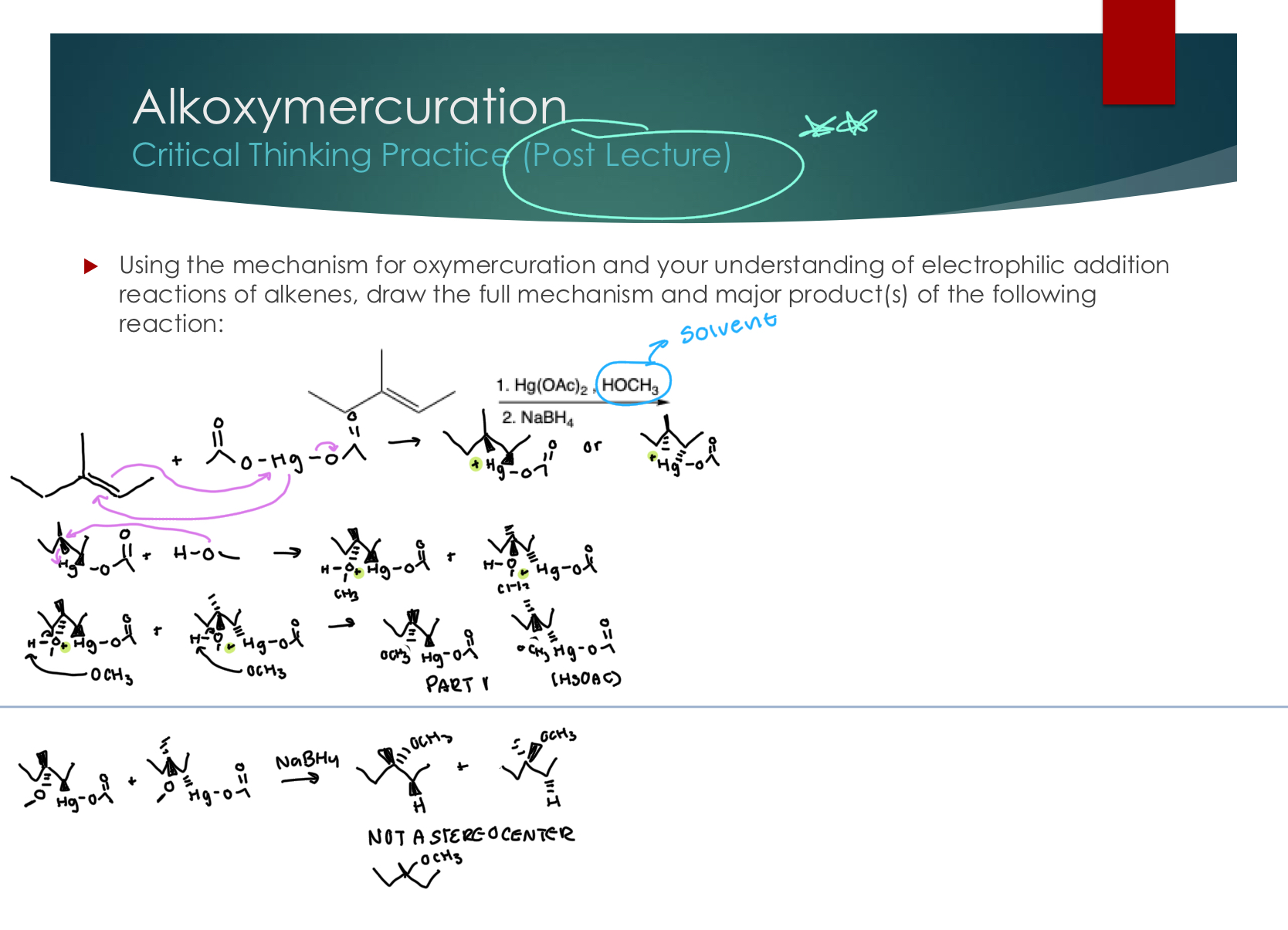
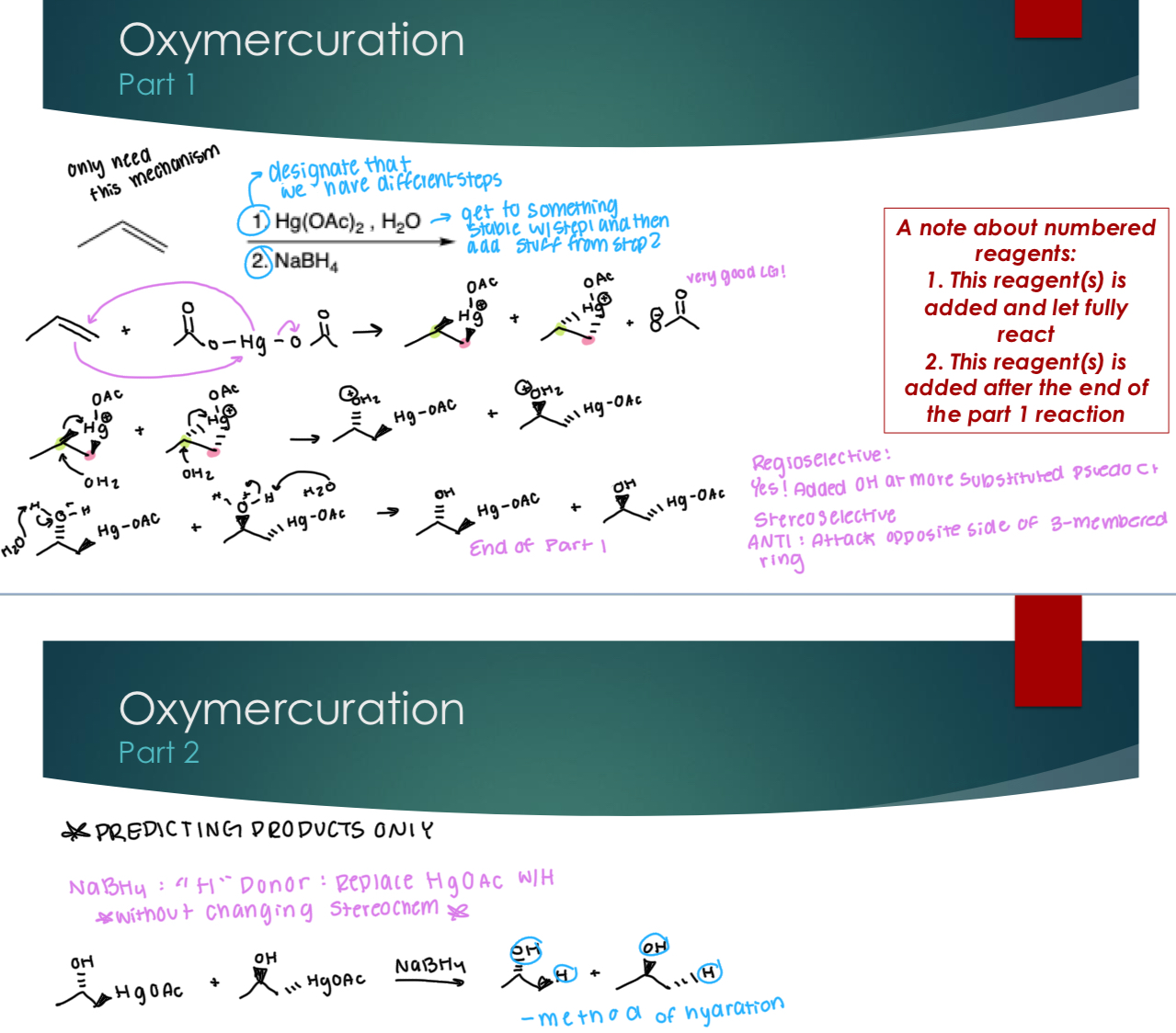
Oxymercuration
A note about numbered reagents:
1. This reagent(s) is added and let fully react
2. This reagent(s) is added after the end of the part 1 reaction
PART 1
Stereoselectivity: Yes! ANTI: attack opposite sides of the 3-membered ring because it creates the more stable product.
Regioselectivity: Yes! Added OH at more substituted pseudo C+ and the “H” at less subsitiuted Carbon
PART 2 PREDICTING PRODUCTS ONLY
NaBH4: “H” Donor: Replace HgOAc with H without changing the stereochemistry
Alkoxymercuration