haemostasis/the coagulation cascade
1/42
Earn XP
Description and Tags
week 3 block 2 ctb
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
43 Terms
haemostasis
the process to limit blood loss from damaged vessels
the precisely orchestrated series of regulatory processes that culminate in the formation of a blood clot that limits bleeding from an injured vessel
haemostasis allows:
blood to be in a fluid state in normal vessels
formation of a localised haemostatic clot at sites of vascular injury
prevents haemorrhage
achieved by a balance between a procoagulant and anticoagulant reactions that occur continuously in the blood
physiological processes
coagulation: process of formation of a haemostatic plug (clot)
fibrinolysis: the process of the breakdown of fibrin within a haemostatic plug (clot)
pathological processes
haemorrhage: the extravasation of blood into the extravascular space
thrombosis: the formation of a solid mass of blood products in a vessel lumen
main components of haemostasis
vascular wall (endothelium and subendothelial structures)
platelets
coagulation cascade
division of blood vessel wall
tunica intima (next to lumen)
tunica media
tunica adventitia
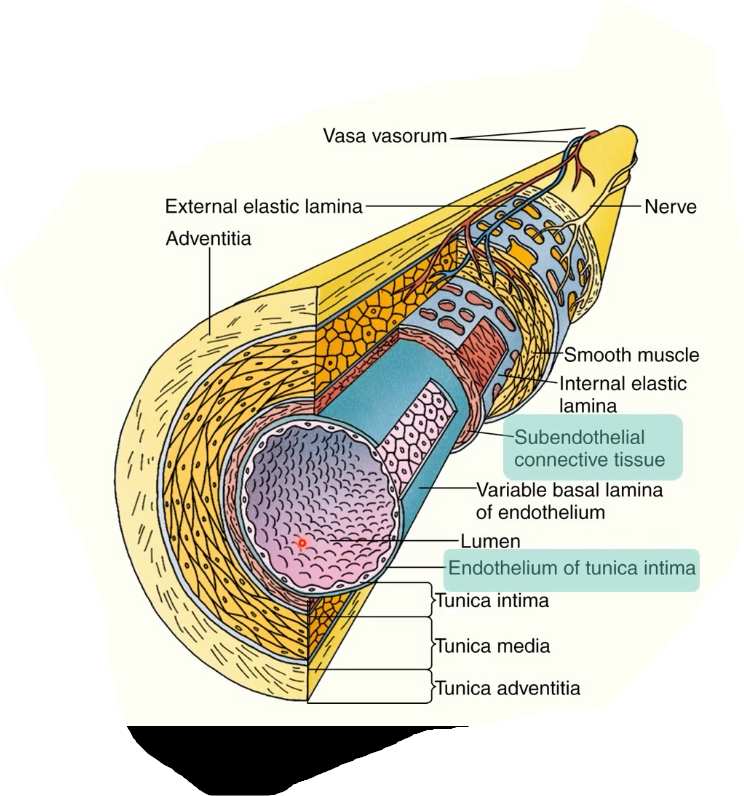
subendothelial connective tissue under tunica intima
endothelium= single layer of squamous cells lining the lumen of the vessel
subendothelium = layer of connective tissue containing collagen
role of endothelium
antiplatelet
inhibits platelets/coagulation cascade and promotes the breakdown of clots
anticoagulant
fibrinolytic
expresses factors to prevent thrombosis in undamaged vessels and limit clot formation to site of vascular injury
act as a barrier between procoagulant subendothelium and coagulation factors in the blood
damage to endothelial cells cause them to release factors which promote coagulation and exposes the subendothelium
role of platelets
form the primary haemostatic plug
provide a surface for recruitment and concentration of coagulation factors and acts as a catalytic membrane
steps in haemostasis
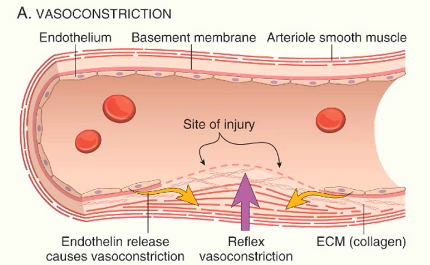
vasoconstriction
primary haemostasis
secondary haemostasis
clot stabilisation and resorption
vasoconstriction
mediated by reflex neurogenic mechanisms and release of endothelin from endothelial cells
minimises blood loss
maximises interactions between platelets, clotting factors vessel wall
primary platelet plug
3 stages to the formation of the primary platelet plug:
platelet adhesion
platelet activation
platelet aggregation
platelet adhesion
blood contains circulating platelets
endothelium has antiplatelet properties normally, but damage to the vessel wall exposes the subendothelium
von Willebrand factor circulates in blood/is released by endothelial cells
acts as an anchor between the first platelets arrive at the site of the injury and damaged the vessel wall
vWf binds collagen
platelets binds to vWf
monolayer of platelets form (within 1-2)
plts then bind directly to collagen after transient tethering
platelet activation
plts are activated once they bind to the subendothelium and change shape
become more spherical and develop projections in their cytoplasm (makes them look spiky)
change in shape of GPIIb/IIIa (receptors on the plt surface)
allows plt-plt interactions to takes place
target for antiplatelet drugs
activated platelets release ADP and thromboxane A2: platelet release reaction
helps promote haemostasis and activates further platelet activation
can be modulated by antiplatelet drugs
plts secrete fibrinogen (acts as a cross-bridge) and allows plts to bind to each other via their GPIIb/IIIa receptors and vWf receptors
serotonin promotes vasoconstriction
calcium ions released (used in secondary haemostasis)
platelet aggregation
further platelets activated
activated platelets bind via their GPIIb/IIIa receptors to other platelets
form fibrinogen cross-bridges
more and more platelets are recruited to the area and activated and get stuck together
cross linking between platelets: platelet aggregation
mass of platelets form a primary haemostatic plug
primary haemostatic plug seals vessel wall and stops bleeding
stabilisation and reinforcement of the plug is necessary
secondary haemostasis: stabilisation and reinforcement of plug
acts on the fibrinogen cross-bridges between platelets
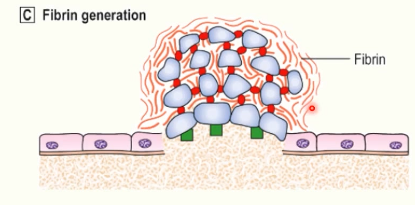
fibrin generation
damage to vessel wall → exposure of tissue factor on subendothelial cells
TF binds and activates factor VII → coagulation (clotting) cascade
thrombin generated → cleaves fibrinogen into fibrin
fibrin polymerises into long chains
consolidates primary platelet plug and forms a secondary haemostatic plug
clotting/coagulation cascade
proteolytic cleavage of pro-enzymes to active enzymes
amplification system
proteins involved are called clotting/coagulation factors
goal: produce thrombin which converts fibrinogen to fibrin, stabilising the clot (secondary haemostasis)
coagulation cascade requirements
coagulation factors (pro-enzymes) → activated coagulation factors (enzymes)
factors XII, XI, IX, X, VII and prothrombin (factor II)
active form indicated by ‘a’ e.g: FXIa
cofactors (reaction accelerators)
factors V and VIII
negatively charged phospholipid surface
activated plts
Ca2+ ions
vitamin K
factors VII, IX, X and prothrombin require are dependent on vit K for correct production
coagulation pathways
traditional pathways:
extrinsic
intrinsic
(separated by laboratory assays)
both lead to final common pathway
extrinsic pathway
prothrombin time (PT)
in lab, initiated by adding tissue factor, phospholipid, calcium to a plasma sample and recording the time for a fibrin clot to form
called extrinsic factor as the blood isn’t normally exposed to TF (as it’s in the subendothelium)
TF activates factor VII and forms a complex with calcium ions
this complex activates factor IX in intrinsic pathway
and factor X starts the final common pathway
the amount of factor Xa produced in extrinsic pathway is small compared to the factor Xa produced in intrinsic pathway
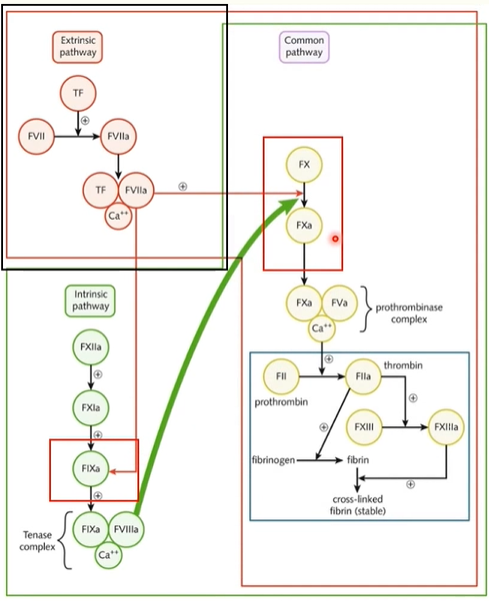
intrinsic pathway
clinically measured as activated partial thromboplastin time (aPTT)
initiated in lab by adding negatively charged particle, phospolipids and calcium to a plasma sample and recording the time for a fibrin clot to form
intrinsic pathway initiated when factor XII comes into contact with a negatively charged surface
(this would be the cell membrane of an activated plt in the body)
intrinsic pathway cont
factor XII activation
which cleaves factor XI into XIa
this then cleaves factor IX into factor IXa
activated factor IX forms a complex with factor VIIIa and calcium ions: the tenase complex
tenase complex is powerful activatory for factor X (tenase is an enzyme for factor X)
therefore large amounts of factor X are produced
final common pathway
begins with activation of factor X either by tenase complex from intrinsic pathway or tissue factor VIIa calcium complex from extrinsic pathway
factor Xa forms a complex with factor Va and calciu ions called: prothrombinase complex
prothrombinase cleaves prothrombin to thrombin
actions of thrombin
conversion of fibrinogen to fibrin
amplifies coagulation process by further activating:
FXI
FVIII
FV
activates FXIII → covalently cross linking fibrin polymers which stabilises the secondary haemostatic plug
further platelet activation
proinflammatory effects: contributes to tissue repair and angiogenesis (process of forming new blood vessels from pre-existing ones)
anticoagulant effects: when interacting with normal endothelium → helps limit clots to the site of the injury
in vivo pathway
initiation phase
amplification phase
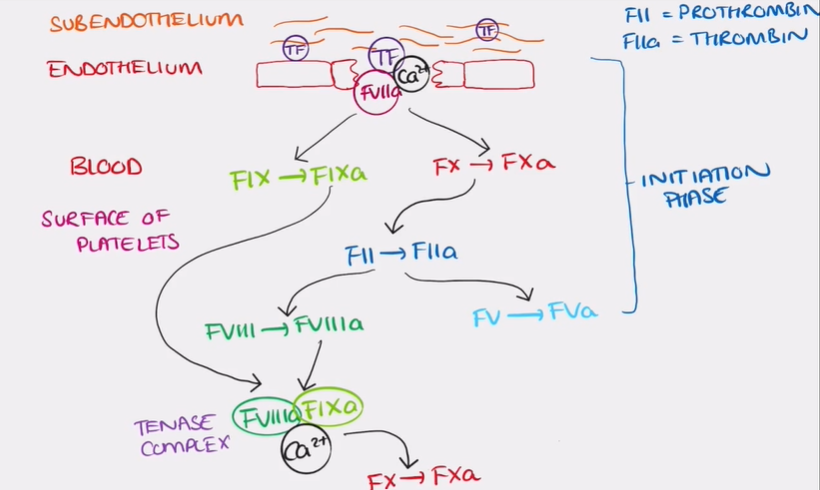
initiation phase of in vivo pathway
exposure of TF in the sub endothelium which then activates and binds to TF VII
form a complex with calcium ions and does 2 functions:
activates TF IX
activates a small amount of factor X
only a small amount of factor Xa is produced
amplification phase of in vivo pathway
the surface of an activated plt acts as a catalyst for the conversion of a small amount of prothrombin to thrombin by Xa on its own
(prothrombin= factor II and thrombin = factor IIa)
thrombin then activates factor VIII to factor VIIIa and factor V to factor Va
first phase of in vivo pathway = initiation phase
factor VIIIa and factor IXa form the tenase complex
potent activator for factor X than TF VIIa-calcium complex
large amount of factor X is produced and factor Xa and forms a complex with factor Va (prothrombinase complex)#
thrombin converts fibrinogen → fibrin
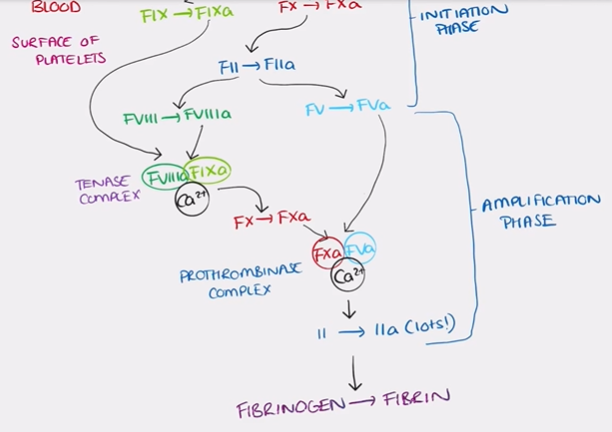
control mechanisms are needed:
to ensure restriction of coagulation to the site of injury
to prevent spontaneous activation of coagulation in the absence of injury
factors that limit coagulation
dilution: washes away coagulation factors
need for a negatively charged surface provided by activated platelets
adjacent intact endothelium: antiplatelet, anticoagulant, fibrinolytic
circulating inhibitors:
antithrombin III: actively augmented by heparin-like molecules on intact endothelium → inhibits thrombin, FIXa, FXa, FXIa and FXIIa
fibrinolytic cascade
limits the size of clots amd contributes to their breakdown
heparin
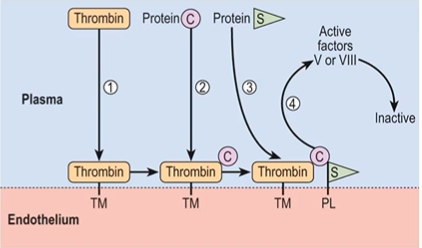
actions of adjacent intact endothelium
physical separation of blood from subendothelium
platelet inhibitory factors
fibrinolytic effects
tissue plasminogen activator (t-PA)
anticoagulant effects
TF pathway inhibitor: inhibits the TF-FVIIa- Ca2+ complexes
thrombomodulin and endothelial protein C receptor: activates protein C, protein C/S complex inhibits factors Va and VIIIa
heparin-like molecules: binds and activates antithrombin IIIfibrinolytic effects
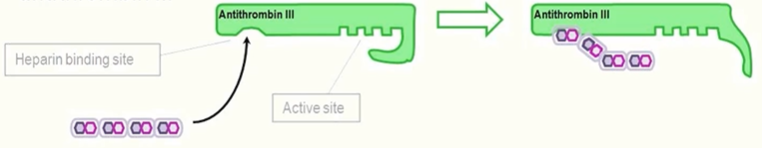
heparin MOA
bnds reversibly to antirhombin III and enchances inactivation of thrombin and FXa
activates antithrombin III by inducing a conformational change that opens up antithrombin III
inhibits thrombosis (low dose)
prevents progression of existing clots (higher dose)
used in prophylaxis and treatment of venous thromboembolism
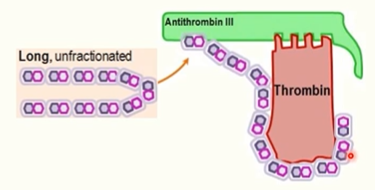
unfractioned heparin
inactivates both FXa and thrombin
unfractioned heparin contains longer chains that can also stabilise antithrombin III complexes with thrombin, leading to thrombin inactivatioN
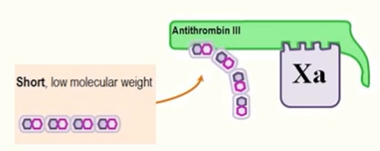
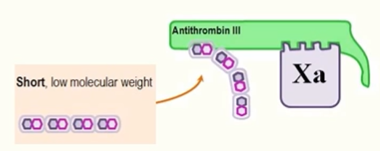
low molecular weight heparin
e.g: dalteparin
primarily inactivates FXa
heparin-activated antithrombin III binds FXa directly via the open active site and inactivates FXa
preferred to UFH in pts (except in severe renal failure)
because LMW heparin has a more predictable response
more favourable side effects profile
doesn’t need routine plasma monitoring or dose adjustments
Fondaparinux
synthetic pentasaccharide (similar structure to heparin)
binds irreversibly to antithrombin III
enhances antithrombin III’s ability to inhibit factor Xa but doesn’t inactivate thrombin (due to shorter chain length)
treatment indications:
VTE prophylaxis for surgical pts
treatment of unstable angina and NSTEMI
warfarin
affects vitamin K metabolism
inhibits synthesis of vitamin K-dependent coagulation factors (factors VII, IX, X and prothrombin)
prophylaxis and treatment of venous thromboembolism and prevention of ischaemic stroke in AF
direct oral anticoagulants (DOACs)
e.g: dabigatran: competitive, reversible thrombin inhibitor
has a similar/greater efficacy than warfarin, fewer drug interactions and no monitoring at standard doses required
clot stabilisation and resorption
FXIIIa mediates formation of covalent cross-links between fibrin polymers
polymerised fibrin and platelet aggregates undergo contraction to form a permanent plug
counter-regulatory mechanisms limit the coagulation to the site of the injury
clot reabsorption and tissue repair
involves the fibrinolytic system (mechanism by which clots are broken down)
important to limit clot size and contributing to their dissolution when they’re no longer needed
D-dimer
degradation product of crosslinked fibrin (by factor XIIIa) and indicates ongoing activaition of haematostatic system
non-specific
can be raise in many physiological/pathological condition
infection
inflammation
malignancy
pregnancy
negative test can be useful but +ve is less useful
fibrinolytic system
enables clot breakdown
removes and limits clot size
inactive circulating plasminogen is converted to plasmin
converted either by FXII-dependent pathway or by plasminogen activators (tissue plasminogen activator t-PA)
plasmin breakd down fibrin polymers
antifibrinolytic factors oppose fibrinolysis
haemorrhage
extravasation of blood into extravascular space due to blood vessel damage
tissues
body cavities
out of the body
can result in:
purpura (visible haemorrhage into skin/mucous membrane) → non blanching
ecchymoses (bruises)
larger, often associated with trauma
petichiae (very small haemorrhages, less than 3mm)
mechanisms of haemorrhage
damage to blood vessel
trauma
atherosclerosis
inflammatory/neoplastic erosion
chronically congested tissues
defective haemostasis: haemorrhagic diatheses
inherited (haemophilia A/factor VIII deficiency)
acquired (disseminated intravascular coagulation DIC)
factors affecting clinical significance of haemorrhage
volume of blood loss
rate of blood loss
medical fitness pre blood loss
site of bleeding
chronic/recurrent external blood loss