11 Retinitis Pigmentosa
1/30
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
31 Terms
What is retinitis Pigmentosa?
A collection of diseases that share similarities in phenotype. They result in the degeneration of photoreceptors and the RPE.
What gene mutations are linked to RP?
Close to 100 with many modes of inheritance:
autosomal dominant ~30-40%
Autosomal recessive ~50-60%
X-linked ~5-10%
~50% of diagnosed subjects, there is no known prior family history (presumed autosomal recessive)
What are the symptoms of RP?
Symptoms are variable as RP is a colleciton of diseases, most RP affect rods before cones, but some variants affect cones first.
Most common presenting symptom is nyctalopia (diff seeing at night) that first presents in late childhood to early adulthood
Visual field loss: pt notices loss of peripheral vision
In cone-rod RP, presenting symptoms can instead be acuity loss, photophobia, photopsia, and color vision concerns
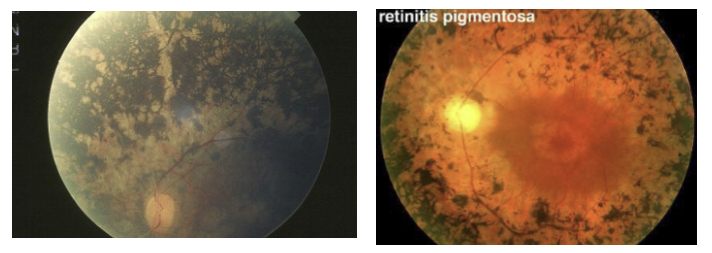
What are common fundus findings for RP?
Pigment clumping in peripheral retina that has “bone spicule” appearance is a classic sign. Also attenuated retinal arteries.
What is the most common gene defect in autosomal dominant retinitis pigmentosa (adRP), and what is the core pathogenic mechanism?
Most common mutation: Rhodopsin (RHO) gene
Mechanism: Misfolded/aberrant rhodopsin accumulates → toxic to rod photoreceptors
Leads to early rod degeneration → nyctalopia (night blindness)
Why do cones and the retinal pigment epithelium (RPE) eventually degenerate in rhodopsin‑mediated adRP?
Secondary degeneration due to rod loss altering retinal environment
Loss of trophic support, increased oxidative stress, and metabolic strain on RPE
Cones die later → progressive loss of peripheral and central vision
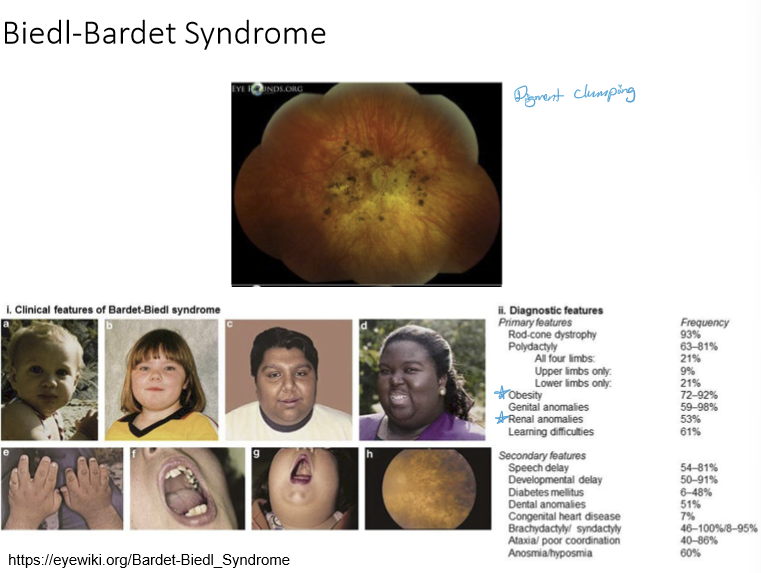
What is Bardet-Biedl syndrome?
A rare, inherited ciliopathy that affects multiple body systems. Pt typically present with obesity and renal anomalies.
What is the underlying cause of Usher syndrome, and what two organ systems are primarily affected?
• Caused by mutations in genes required for development/function of the inner ear and retina
• Leads to a retinal phenotype similar to RP + congenital deafness
• Dual involvement: hearing + vision
What is the key cellular structure implicated in the pathophysiology of Usher syndrome?
Involves cilia-related proteins
Cilia connect the outer and inner segments of photoreceptors
Cilia present in hair cells of inner ear
What other ciliopathy is relevant when thinking about the mechanism of Usher syndrome?
Bardet–Biedl Syndrome (BBS) also involves cilia dysfunction
What does PDE6 gene code for?
One type of phosphodiesterase
What does RPE65 gene code for?
protein involved in recycling chromophores
What is CNGA/B gene code for?
Codes for subunitsd in cGMP-gated channels
Can RP affect unilaterally?
Yes, very rarely can affect only 1 eye instead of bilaterally.
How does dark adaptation testing help in diagnosing and monitoring retinitis pigmentosa (RP)?
Dark adaptation can detect early rod dysfuction
Shows abnormalities in the rod-cone break (delayed) and threshold levels (rod plateau elevated)
Useful for disease monitoring and progression and assessment
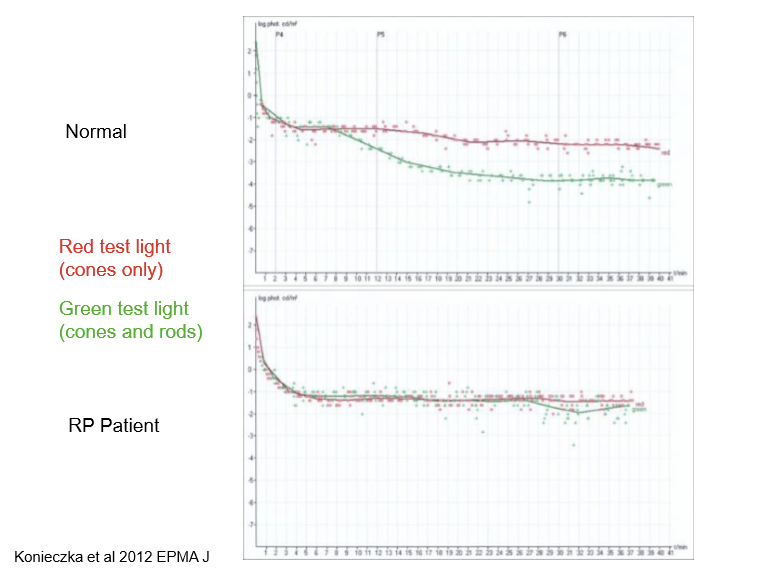
What happens to the rod–cone break and rod plateau in RP during dark adaptation?
• Rod–cone break is delayed, similar to AMD
• Rod plateau threshold is significantly elevated early → hallmark of early rod degeneration
• Reflects primary involvement of rods in most RP subtypes
How are cone thresholds affected in RP as the disease progresses?
• As cones become involved later, cone plateau thresholds become elevated
• Progression from rod‑predominant dysfunction → combined rod + cone impairment
• Leads to worsening photopic sensitivity over time
What is Leber’s congenital amaurosis (LCA), and how is it distinguished from retinitis pigmentosa (RP)?
• Considered by some a severe, early‑onset form of RP, by others a separate entity
• Defined by vision loss at birth or within the first year of life
• Represents congenital photoreceptor dysfunction rather than later‑onset RP
What genetic defect defines LCA type 2, and what is the underlying mechanism?
• RPE65 gene mutation → LCA type 2
• RPE65 normally converts all‑trans‑retinyl esters → 11‑cis‑retinal in the visual cycle
• Loss of RPE65 → failure of phototransduction → severe early blindness
What is the current approved therapy for RPE65‑associated LCA (type 2), and how is it delivered?
• Gene therapy: AAV vector carrying functional RPE65
• Delivered via subretinal injection
• Restores the visual cycle → improves functional vision in eligible patients
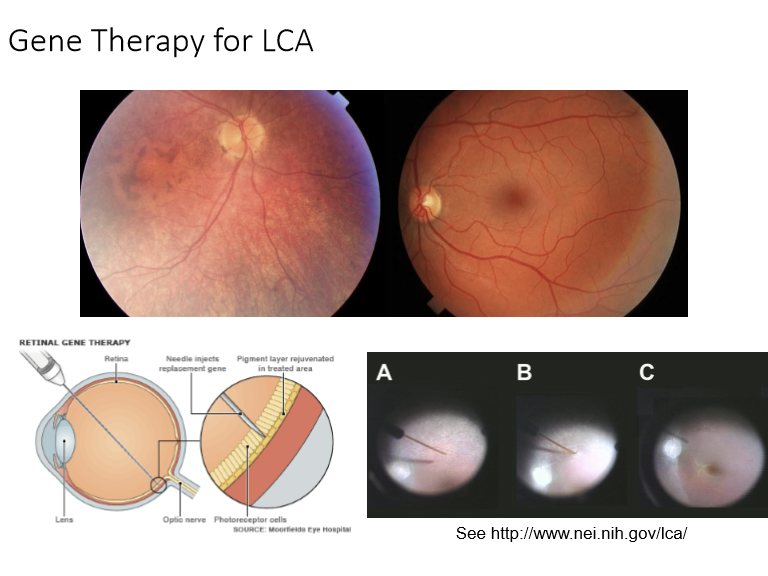
What major milestones illustrate the multi‑decade path from RPE65 discovery to effective human gene therapy for Leber Congenital Amaurosis?
• 1993–1998 (Foundational discoveries):
– RPE65 cloned (1993) → gene mutations linked to LCA (1997)
– Knockout mouse created and shown to disrupt vitamin A metabolism (1997–1998)
– RPE65 mutation identified in blind Briard dogs (1998)
• 2001–2005 (Preclinical breakthroughs):
– Gene therapy restores vision in Briard dogs (2001)
– Vision improvement shown to persist >4 years (2005)
• 2007–2009 (Translation to humans):
– Human clinical trial begins using rAAV2‑RPE65 (2007)
– First cohort completed (2008)
– One‑year results show sustained visual gains and safety (2009)
What are the key clinical findings and current status of gene therapy for retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA)?
• Safety: Clinical trials show gene therapy for LCA is safe in adults and children with no severe adverse effects.
• Efficacy: Leads to modest improvement in visual acuity and expansion of kinetic visual fields.
• Current status: Now FDA‑approved and available (RPE65 gene therapy).
• Pipeline: Additional gene therapies targeting non‑RPE65 causes of RP/LCA are currently in ongoing clinical trials.
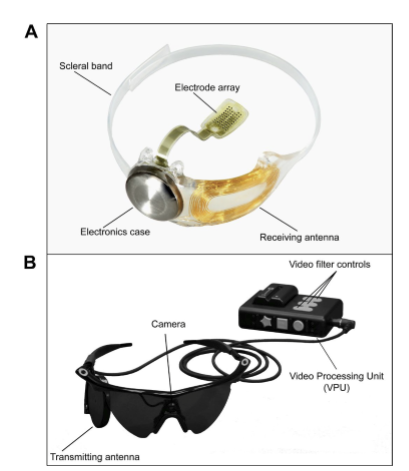
What is the Argus II retinal prosthesis, and what are its key clinical outcomes and safety findings?
• Status: Approved in Europe and received FDA approval in 2013.
• Components: External camera → video processing unit → transmitting antenna; internal receiving antenna + electronics case + scleral‑fixated electrode array.
• Clinical trial: 30 patients implanted; after 5 years, 18/30 had no device‑ or surgery‑related serious adverse events.
• Efficacy: Improvements were modest—patients could better locate objects (e.g., pointing to squares) and perceive direction of motion.
• Purpose: Provides basic visual perception for individuals with late‑stage RP.
What is the core mechanism behind how stem cell therapies may help patients with retinitis pigmentosa (RP)?
Two mechanisms:
1. Replace dysfunctional retinal cells (RPE + photoreceptors) with healthy stem‑derived cells
2. Rescue surviving cells by releasing trophic factors
• Goal: Delay degeneration and potentially restore vision in RP
Why are stem cell therapies particularly promising for RP cases involving RPE‑related gene defects?
• RPE dysfunction is central to many RP variants
• Pluripotent stem cells can replace damaged RPE + photoreceptors, restoring support for photoreceptor survival
• Provides a gene‑agnostic treatment option: useful when no single‑gene therapy exists
What have animal studies shown regarding stem cell therapy for RP?
Animal studies have provided positive results for stem cell therapies with adverse side effects being small. Currently, there are clinical trials on humans.
What is the rationale for using optogenetics in diseases like retinitis pigmentosa (RP)?
• In RP, the outer retina (photoreceptors) degenerates first
• The inner retina (bipolar cells, RGCs) often remains relatively intact
• Optogenetics aims to restore light sensitivity by making inner retinal neurons photosensitive, bypassing lost photoreceptors
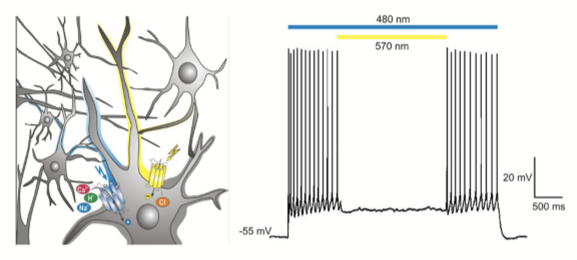
How does optogenetics make inner retinal neurons (typically RGCs) responsive to light?
• Inject rhodopsin‑like, light‑activated ion channels into RGCs
• Short‑wavelength light → opens channels for positive ions (depolarizing)
• Long‑wavelength light → opens channels for negative ions (hyperpolarizing)
• Recreates a basic “photoreceptor‑like” response in cells that normally don’t detect light
What is the current research stage of optogenetics for retinal diseases?
• Still experimental
• Demonstrated in animal models only
• Not yet in human clinical trials
What major therapeutic strategies are being developed to restore or preserve vision in retinitis pigmentosa (RP)?
Restorative therapies:
Gene therapy (correct defective genes)
Neuroprostheses (e.g., retinal implants)
Stem cell therapy (replace/repair RPE + photoreceptors)
Disease-modifying therapies
Neuroprotection to slow degeneration
What supportive, real‑world intervention is essential for patients with RP, regardless of disease stage?
• Low vision assessment and rehabilitation
• Optimizes remaining visual function, safety, and independence
• Crucial for quality of life, even as disease progresses and while emerging treatments are still in development