Looks like no one added any tags here yet for you.
Most common cancers
- Breast/prostate
- Lung and bronchus —> causes the most deaths
- Colorectal
Tumorigenesis
- Controlled by:
Proto-oncogenes which turn into oncogenes:
Proto-oncogenes control the normal cell life. They code for proteins that help to regulate cell growth and differentiation. They are involved in signal transduction and execution of mitogenic signals. Examples include ras, wnt, myc, erk, and trk.
Oncogenes are formed from proto-oncogenes via spontaneous point mutations, inherited germline mutations, misregulated protein expression, or chromosomal translocation.
Tumor suppressor genes are inhibited:
Can cause tumors
Includes p53, p21, plNK4A, and Rb
Cell cycle phases
- Quiescent/senescent state:
G0 —> resting phase where the cell has left the cycle and has stopped dividing
- Interphase state:
G1 —> cells increase in size and enzyme production occurs in preparation for de novo nucleic acid synthesis. Ensures that everything is ready for DNA synthesis.
S —> DNA replication occurs
G2 —> during the gap between DNA synthesis and mitosis, the cell will continue to grow. Ensures that everything is ready to enter the mitosis phase.
- Cell division state:
M —> cell growth stops at this stage and cellular energy is focused on the orderly division into 2 daughter cells.
Metastatic cascade
- Metastasis —> malignant cells leave the parent tumor, migrant to distant sites, and invade new tissue
1. Cells detach from the basement membrane
2. Invasion of the capillary wall
3. Travel through the blood stream
4. Adhere to the capillary wall
5. Escape from blood vessels (extravasation)
6. Proliferate to form metastases
Classification of cancers
- Carcinoma —> derived from epithelial cells including breast, prostate, lung, pancreas, and colon cancers
- Sarcoma —> derived from connective tissue that develop from cells originating in mesenchymal cells outside the bone marrow
- Lymphoma and leukemia —> derived from hematopoietic cells that leave the marrow and tend to mature in lymph nodes and the blood
- Germ cell —> derived from pluripotent cells including testicular and ovarian cancers
- Blastoma —> derived from immature, undifferentiated cells or embryonic tissue
Leukemia classifications
- Myelogeneous leukemia:
Affects granulocytes (basophils, neutrophils, and eosinophils) and monocytes
Includes acute myelogeneous luekemia (AML) and chronic myelogeneous leukemia (CML)
- Lymphocytic leukemia:
Affects lymphocytes
Includes acute lymphocytic leukemia (ALL) and chronic lymphocytic leukemia (CLL)
Platinating/alkylating agents
- 2 classes:
Platinum-based
Carbon-based
- Cytotoxic mechanism:
Not cell cycle specific
More toxic to G1 and S phases
Form stable bonds with the purine bases of DNA
- Bifunctional alkylating agents:
Form 2 bonds to cross-link DNA
Includes platinating agents, nitrogen mustards, etc.
- Monofunctional alkylating agents:
Includes nitrosoureas, dacarbazine, procarbazine, and temozolomide
- 2 kinds of DNA cross-linking:
Intrastrand (95% of the total DNA adduct) and interstrand (5% of the total DNA adduct)
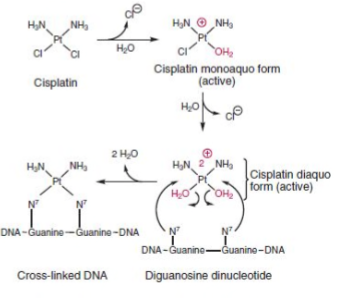
- Platinating agents attach to N7 of adenine and guanine
- DNA repair mechanisms are usually unable to correct the damage —> cell cycle arrest and apoptosis
- All currently marked platinating agents are Pt(II) complexes with a square planar geometry in the cis-configuration.
Side effects of platinating agents
- Severe emetogens
- Myelosuppression which can lead to reduced RBC, WBC, and platelet count
- Ototoxicity which can lead to irreversible hearing loss
- Cumulative nephrotoxicity associated with cisplatin:
Patients should be hydrated with a Cl- solution (NaCl) to reduce the concentration of the active drug in the kidney
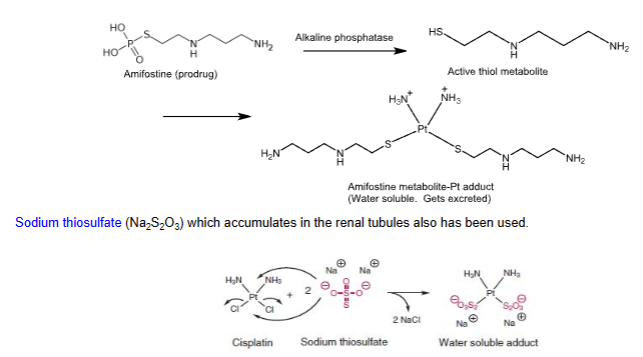
Sodium thiosulfate (Na2S2O3), which accumulates in the renal tubules also has been used
Mannitol can also be used to promote continuous excretion of the drug and its hydrated form
- Anaphylactic-like reactions including facial edema, bronchoconstriction, tachycardia, and hypotension may occur within minutes of cisplatin administration:
Epinephrine, corticosteroids, and antihistamines can be used to alleviate symptoms.
- Extravasation risk
Chemoprotectants
- Used to mitigate the side effects of platinating therapy
- Highly water soluble complexes that get excreted from the kidney
- One example is amifostine
Resistance to platinating therapy
- Compromised carrier-mediated cellular transport by copper-transporting protein (CTR1)
- Intracellular inactivation through drug trapping vesicles
- Inactivation through conjugation -SH (cysteine, GSH, etc.)
- Increased DNA repair by nucleotide-excision repair proteins (NERs)
- Testicular cancer is particularly responsive to cisplatin due to their inherent deficiency in DNA repair processes
Cisplatin
- Platinating agent
- Used to treat metastatic testicular tumors and ovarian tumors in combination with other anticancer agents or post surgery/radiotherapy
- Used also as a single agent to treat advanced bladder cancer not amenable to surgery or radiotherapy
- IV
- Forms a reactive intermediate known as the active diaquo form, which is mediated by a hydration reaction and is more active than cisplatin itself.
- Ensure patients are hydrated for 24 hours with chloride containing solutions to increase urine output because the drug is nephrotoxic.
- Vesicant so be aware of the extravasation risk
- No established antidote and dialysis is ineffective due to high plasma protein binding
Carboplatin
- Platinating agent
- Used to treat ovarian carcinomas in combination with cyclophosphamide
- Has the same reactive intermediate as cisplatin
- Less potent and has a milder side effect profile than cisplatin because the diaquo intermediate is formed 10x slower
- Plasma half-life is 3 hours
Oxaliplatin
- Platinating agent
- Used to treat stage III colon cancer and advanced colorectal cancer alone or in combination with 5-fluorouracil
- Does not have the same active intermediate as cisplatin
- (S,S)-enantiomer is inactive
- Less resistance and better safety than cisplatin and carboplatin
- The conformation of its DNA adduct is not recognized by mismatch repair proteins (MMRs)
- Less dependent on CTR1 active transport proteins for intracellular access compared to cisplatin
- High Vd of 440L —> highly bound to tissues
- Can cause pulmonary fibrosis and life-threatening peripheral sensory neuropathies
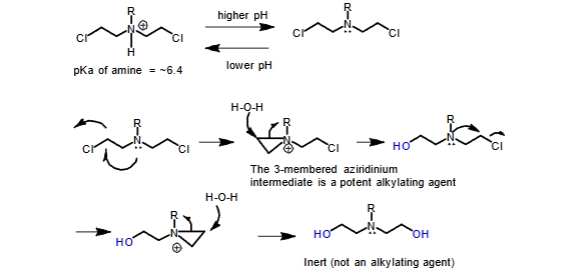
Decomposition of carbon-based alkylating agents in aqueous media to inactive compounds
- The drug is stored at a lower pH where the N will be protonated
- At physiological pH, the unprotonated drug cyclizes to a reactive aziridinium intermediate, which is a potent alkylating agent
Fate of alkylated DNA
Mechlorethamine hydrochloride
- Carbon-based alkylating agent
- Only aliphatic nitrogen mustared currently used in the U.S.
- Used to treat Hodgkin’s disease, CML, and CLL
- IV —> too reactive for oral dosing and needs special handling
- Potent vesicant —> a solution of sodium thiosulfate (Na2O3S2) is used to inactivate the drug where accidental exposure to the skin occurs
- Can cause myelogeneous leukemia with extended use due to its mutagenic/carcinogenic effects on bone marrow stem cells
Melphalan
- Carbon-based alkylating agent
- Aromatic nitrogen mustard —> less basic (pKa = 2.1)
- Less reactive than aliphatic mustards (pKa ~ 6)
- ^^^Higher stability leads to greater distribution into cancer cells and a decreased incidence of severe side effects^^^
- Oral (can be erratic and food impacts absorption) and IV
- Used to treat multiple myeloma and epithelial carcinomas of the ovary
- Less N/V compared to mechlorethamine
- Mutagenic and can induce nonlymphocytic leukemia
- Distributes into body water, so it is more toxic in dehydrated patients
Chlorambucil
- Carbon-based alkylating agent
- Aryl
- Used to treat palliative hematological cancers including CLL, malignant lymphoma, and Hodgkin’s disease
- Good OBA —> negative food effect
- Parent drug is active and undergoes benzylic oxidation to provide an active phenylacetic acid mustard metabolite, which is responsible for some of the observed antineoplastic activity.
- Rapid absorption and clearance in urine
- 99% protein binding
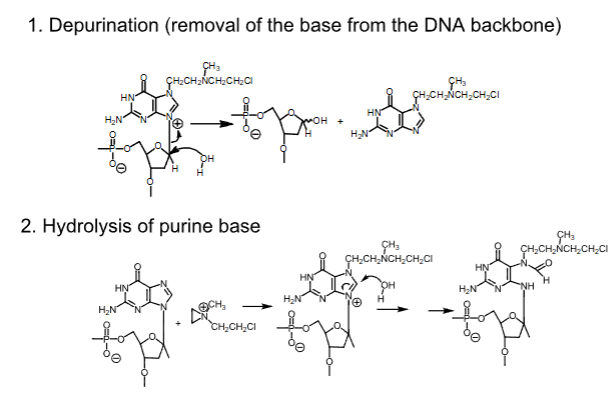
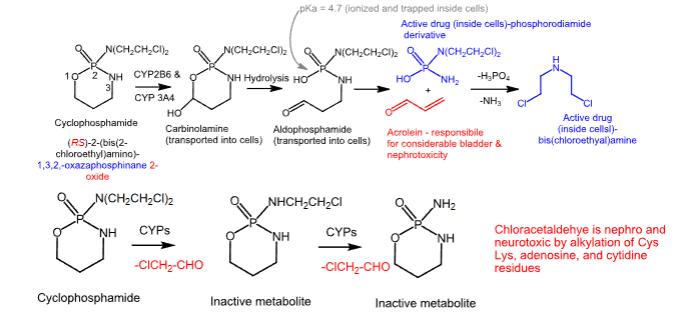
Cyclophosphamide
- Carbon-based alkylating agent
- Prodrug
- Side product of this metabolism is acrolein, which is urotoxic and nephrotoxic
- Chloroacetaldehyde is neurotoxic and nephrotoxic
- Used to treat malignant lymphomas, Hodgkin’s disease, multiple myeloma, and leukemias
- Oral or IV
- F >75%
- Protein binding is low
- Half-life of 3-12 hours
- Biotransformed in the liver
- Can cause emesis, alopecia, and N/V
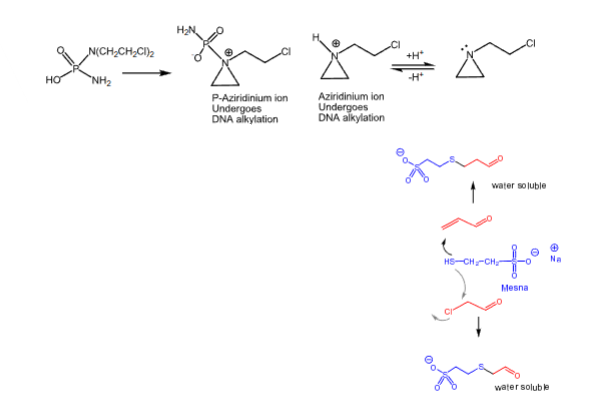
- Mesna can be used as a cytoprotective adjuvant to reduce acrolein-induced toxicities by forming a water-soluble, excretable adduct
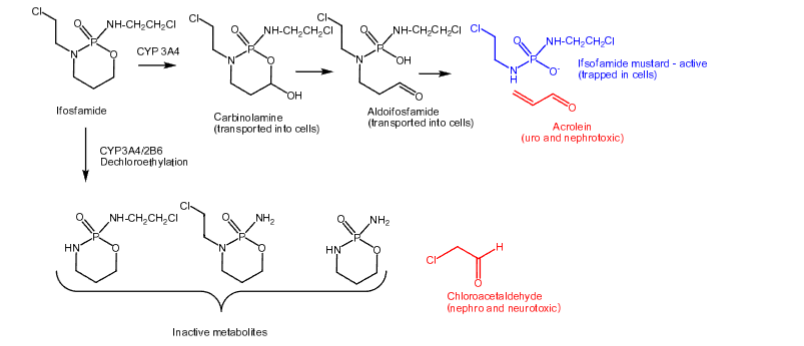
Ifosfamide
- Carbon-based alkylating agent
- Prodrug
- Activated more slowly than cyclophosphamide
- Half-life of 7 hours
- Undergoes N-dechloroethylation like cyclophosphamide to produce inactive metabolites and chloroacetaldehyde (toxicities from above)
- Mesna is used as a uroprotective agent
- Used first-line to treat testicular cancer
- IV
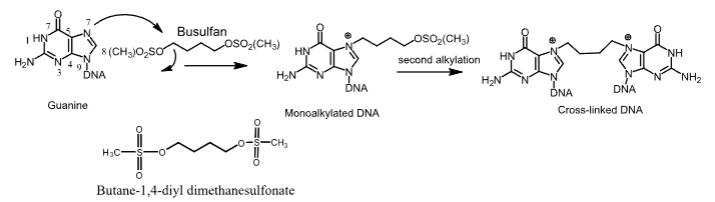
Busulfan
- Carbon-based alkylating agent
- Used to treat CML
- Oral and IV
- Monoalkylated and cross-linked DNA adducts form
- Can cause serious bone-marrow hypoplasia and myelosuppression
- Recovery from busulfan-induced pancytopenia can take up to 2 years
- Crosses the BBB, which can induce seizures
Nitrosoureas
- Unstable compounds that decompose readily in the aqueous environment of the cell to produce highly reactive carbocations, which alkylate DNA.
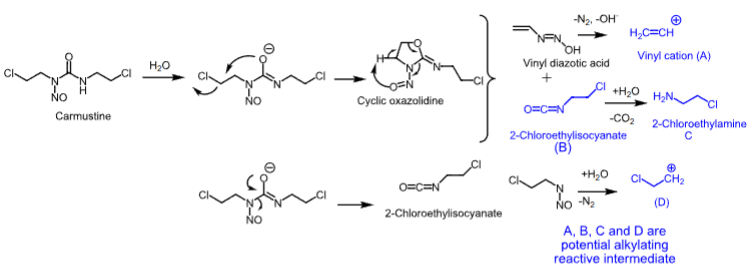
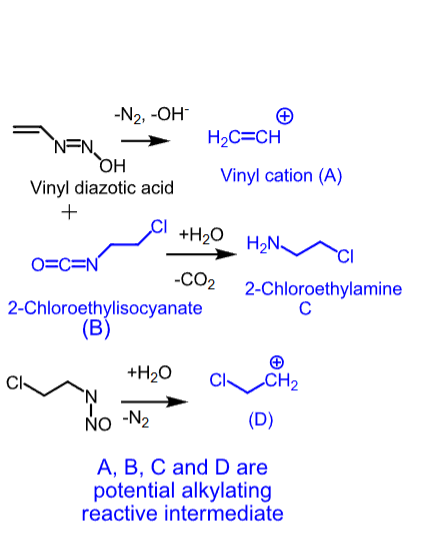
Carmustine
- Nitrosourea
- Used as single or combination therapy to treat brain tumors, multiple myeloma (with prednisone), Hodgkin’s disease, and non-Hodgkin’s lymphomas
- IV or implant (Gliadel wafer)
- Lipid-solube —> crosses the BBB
- Degrades in 15 minutes
- Can cause bone marrow toxicity (delayed myelosuppression), emesis, and acute pulmonary toxicity
Lomustine
- Nitrosourea
- Oral
- Used to treat primary and metastatic brain tumors
- Used to also treat Hodgkin’s disease as secondary therapy in combination with other drugs
- Delayed bone marrow suppression and carcinogenicity
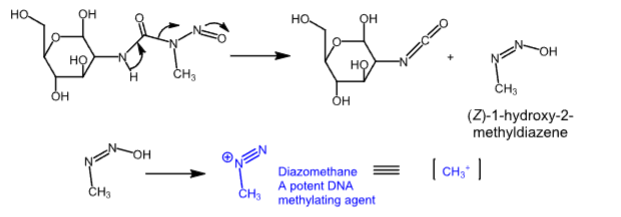
Streptozocin
- Nitrosourea
- Water-solube N-nitrosourea —> islet specific
- Used to treat metastatic islet cell carcinoma (AKA metastatic pancreatic neuroendocrine tumors)
- IV
- Can cause cumulative renal toxicity
- Diazomethane intermediate alkylates DNA
- DNA damage induces the activation of poly ADP-ribosylation, which can lead to diabetes
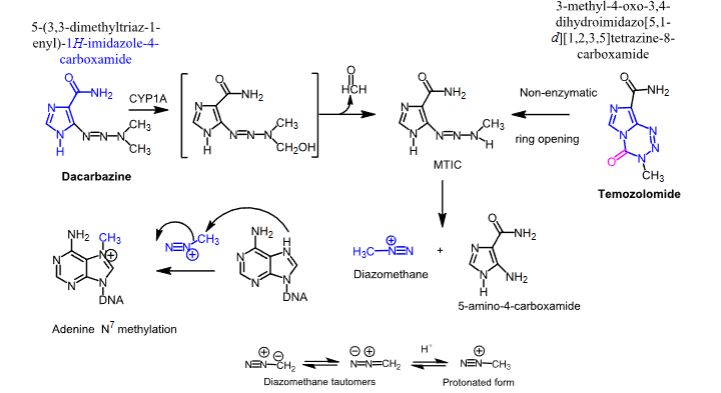
Dacarbazine and temozolomide
- Carbon-based alkylating agents
- Dacarbazine —> undergoes a CYP1A mediated conversion to MTIC
- Temozolomide —> undergoes a hydrolytic ring opening to MTIC
- MTIC undergoes decomposition to the highly reactive diazomethane, which methylates DNA
Dacarbazine
- Carbon-based alkylating agent
- Used to treat Hodgkin’s disease and malignant melanoma
- IV
- Can cause emesis, anorexia, and vomiting
Temozolomide
- Carbon-based alkylating agent
- Used to treat brain cancers, specifically in adults with newly diagnosed glioblastoma multiforme
- Used to also treat refractory anaplastic astrocytoma
- Oral (IV availabe too)
- Can cause neutropenia and thrombocytopenia
Procarbazine and MOPP regimen
- Carbon-based alkylating agent
- Orally active
- Metabolized to methyl radicals that methylate DNA
- Used to treat Hodgkin’s disease
- Administered in a multidrug regimen known as MOPP (developed by the NCI) that is capable of curing almost 70% of patients with advanced-stage Hodgkin lymphoma:
Mechlorethamine (alkylating agent)
Vincristine (Oncovin) —> mitosis inhibitor
Prednisone (steroid)
Procarbazine (DNA methylating agent)
Serious toxicities
Rapid and complete absorption
Crosses the BBB
Metabolized in the liver
Can cause emesis and leucopenia
Strong MAOIs can lead to fatal DDIs
No longer used because of the chance to develop a second cancer within 20 years
Topoisomerase poisons
- Cytotoxic agents
- During DNA replication and transcription, DNA undergoes supercoiling, knotting, and catenation
- Topoisomerase enzymes restore the topology of the overwound DNA by creating transient breaks in the DNA, thereby releasing the strain
- Topoisomerase IIa cleaves both strands of a double-stranded DNA during replication and repairs its supercoiling
- Topoisomerase I functions in the same way, but it cuts and re-ligates a single DNA strand instead of a double DNA strand
Camptothecin
- Topoisomerase I inhibitor
- Binds to the DNA-topoisomerase I complex, preventing the cleaved DNA strand from re-sealing
- Active in the S phase of the cell cycle
- Isolated from the Chinese Xi Shu (happy tree)
- Limited water solubility
- Forms a soluble salt of the carboxylic acid that results from ring opening, but is less active
- Synthetic analogs with basic groups are used to increases its solubility
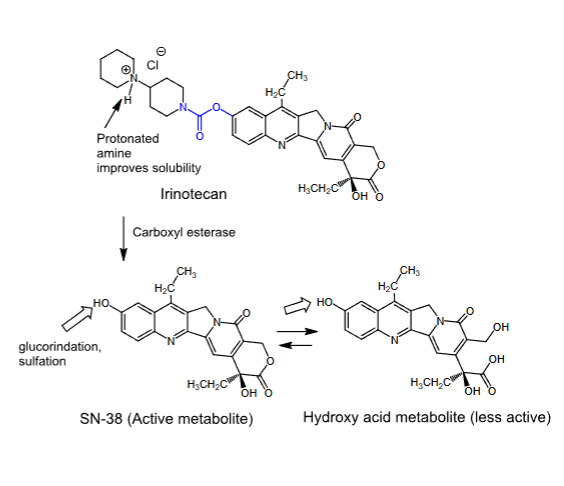
Irinotecan
- Topoisomerase I inhibitor
- Binds to the DNA-topoisomerase I complex, preventing the cleaved DNA strand from re-sealing
- Active in the S phase of the cell cycle
- Synthetic camptothecin analog
- Prodrug
- Incorporates a basic group to improve solubility
- Converted to an active metabolite in the liver
- Used first-line to treat metastatic colorectal cancer in combination with 5-fluorouracil
- IV
- SN-38 is formed slowly from the parent drug:
Metabolized to the less active hydroxy acid metabolite, but preferential protein binding of SN-38 shifts the equilibrium in its favor
Half-life of 11.5 hours
Eliminated by glucuronidation and sulfation at the phenolic hydroxyl group
- Can cause delayed diarrhea due to its anticholinergic effects
- Myelosuppressive —> can cause severe neutropenia
- Those with a deficiency in the active form of UGT1A1 will experience life-threatening toxicities
Topotecan
- Topoisomerase I inhibitor
- Binds to the DNA-topoisomerase I complex, preventing the cleaved DNA strand from re-sealing
- Active in the S phase of the cell cycle
- Oral and IV
- IV formulation is used to treat metastatic ovarian cancer after failed initial chemotherapy, SCLC after failure of first-line therapy, and stage IV-B recurrent/persistent cervical cancer in combination with cisplatin
- Oral formulation is used to treat relapsed SCLC
- Can cause myelosuppression (primarily neutropenia), making its use limited
- Renally cleared
- No known UGT1A1 polymorphism related toxicities
- Has an inactive hydroxy acid metabolite
Podophyllotoxin-derived topoisomerase II inhibitors —> etoposide and teniposide
- Semisynthetic glycosidic derivative of podophyllotoxin, isolated from the dried roots of podophyllum peltatum
- Topoisomerase IIa inhibitors
- Have a 1-epipodophyllotoxin structure with 1-OH group glycosylated
- Once bound to topoisomerase IIa, epipodophyllotoxins allow for cleavage of double stranded DNA, but inhibit the subsequent step of resealing the cleaved DNA
- Cause cell cycle arrest in the S phase
- Used to treat SCLC and refractory testicular cancer
- Oral and IV
- Highly protein bound
- Can cause severe myelosuppression
Antitumor antibiotics
- Cytotoxic agents
- Block DNA replication or transcription
- Topoisomerase IIa poisons —> anthracyclines
- Cytotoxic free radical generators —> bleomycin
- DNA alkylating antibiotics —> mitomycin
- Prevention of transcription by binding to DNA —> dactinomycin
Anthracyclines
- Isolated from streptomyces bacterium
- 2-fold MoAc:
Topoisomerase IIa inhibitor
Hydroxy free radical-induced DNA strand scission
- Can cause cardiotoxicity (hydroxy radical-mediated effect), myelosuppression, N/V, and mucositis
- Used to treat various hematological cancers
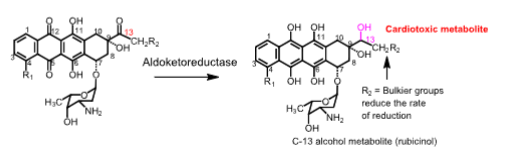
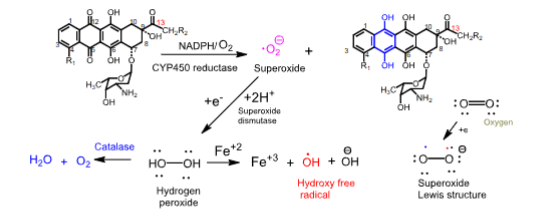
Mechanism of cardiotoxicity of anthracyclines
- When NADPH/CYP450 reductase reduces the quinone ring to a hydroquinone, superoxide radical anions are generated
- Superoxide radicals are reduced to hydrogen peroxide by superoxide dismutase
- Cardiac tissue is vulnerable to damage by anthracyclines because it lacks catalase, which is responsible for forming harmless products like water and O2
- The secondary alcohol metabolites formed by the reduction of the C13 carbonyl group can inhibit Ca2+, Mg2+-ATPase, and Na+/K+-ATPase, leading to chronic cardiac myopathies, resulting in CHF
Doxorubicin
- Anthracycline
- 2-fold MoAc:
Topoisomerase IIa inhibitor
Hydroxy free radical-induced DNA strand scission
- Used to treat hematological cancers (lymphoblastic leukemia and AML) and solid tumors of the breast, ovary, stomach, bladder, and thyroid gland
- A liposomal formulation (Doxil) is used to treat AIDS-related Kaposi sarcoma and organoplatinum-resistant ovarian cancer —> half-life of 55 hours
- IV
Daunorubicin
- Anthracycline
- 2-fold MoAc:
Topoisomerase IIa inhibitor
Hydroxy free radical-induced DNA strand scission
- Used to treat ANL in adults and ALL in children and adults
- IV
Idarubicin
- Anthracycline
- 2-fold MoAc:
Topoisomerase IIa inhibitor
Hydroxy free radical-induced DNA strand scission
- Used to treat AML in adults in combination with other anti-leukemic drugs
- IV
Dexrazoxane
- Chemoprotectant for anthracycline toxicity
- Antioxidant
- Prodrug
- Chelates iron, which is needed for the hydroxy radical forming Fenton reaction
- Penetrates cardiac tissue where it gets hydrolyzed to amide-carboxylate, which is a strong Fe2+ chelator
Mitoxantron
- Synthetic anthracycline analog
- Topoisomerase IIa inhibitor
- Protonated amines interact with anionic phosphate residues on DNA, leading to a ternary complex of mitoxantron-DNA-topoisomerase IIa
- Reduced cardiac toxicity:
Less prone to NADPH/CYP450 reduction, leading to a lowered chance of hydroxy radical formation
No cardiotoxic oxygen radicals (ROS)
- Has a naphthoquinoxaline active metabolite, which is less toxic than the parent
- Used to treat ANL in combination with other drugs and prostate cancer
- IV
- Can cause moderate N/V and bone marrow suppression
Bleomycin
- Anti-tumor antibiotic
- Natural product isolated from streptomyces verticillus
- Mixture of bleomycin A2 and B2
- Intercalates with DNA and causes DNA destruction via cytotoxic free radicals
- Electron rich disaccahrdie, imidazole, and pyrimidine bind to Fe2+
- BLM-Fe2+ —> BLM-Fe3+-OOH —> OH.
- The hydroxy radical abstracts a proton from the 4’ of DNA-ribose, causing DNA scission
- Used to treat Hodgkin’s and non-Hodgkin’s lymphomas, squamous cell carcinoma, and testicular carcinoma
- IV
- 10% of patients develop pneumonitis —> pulmonary fibrosis
- 1% of patients die
- Does not cause bone marrow suppression
Dactinomycin
- Anti-tumor antibiotic
- Derived from streptomyces parvullus
- Discovered by Selman Waksman at Rutgers
- Cyclic bis-depsipeptide attached to a substituted phenoxazine core
- Binds strongly to DNA and inhibits transcription and replication of DNA
- The strong binding to DNA is due to the planar tricyclic aromatic ring system
- Binds to the guanine portion of DNA, intercalating between guanine and cytosine pairs
- Used to treat Wilms’ tumor, Ewing’s sarcoma, and solid malignancies
- The p-benzoquinoneimine reduction by NADPH/CYP450 reductase generates hydroxy free radicals that break single-stranded DNA
- IV
- Large Vd
- Minimal metabolism
- Toxicity is frequent and severe
- Risk of extravasation
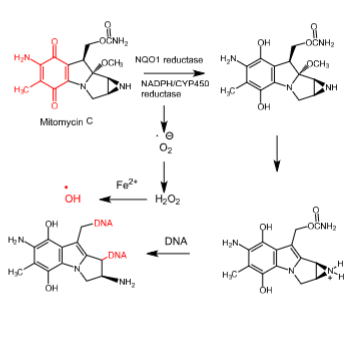
Mitomycin C
- Anti-tumor antibiotic
- Derived from streptomyces caespitosus or streptomyces lavendulae
- 2-fold mechanism of cytotoxicity:
Hydroxy radical generation, which causes DNA cleavage
Direct alkylation of DNA
- Activated through a 2-electron bioreductive process using NADPH/CYP450 reductase and/or NADPH quinone oxidoreductase 1 (NQO1 reductase), an enzyme extensively expressed in many neoplastic cells
- Hydroxyl radicals break single-stranded DNA
- DNA alkylation occurs at 2 sites —> crosslinks DNA
- Used to treat disseminated adenocarcinoma of the stomach or pancreas in combination with other drugs
- Rapidly cleared
- Can cause bone marrow toxicity and cumulative myelosuppression
Antimetabolites
- Cytotoxic agents
- Inhibit de novo DNA synthesis:
Inhibit the enzymes that synthesize nucleotides
Some arrest chain elongation via incorporation into DNA as false substrates
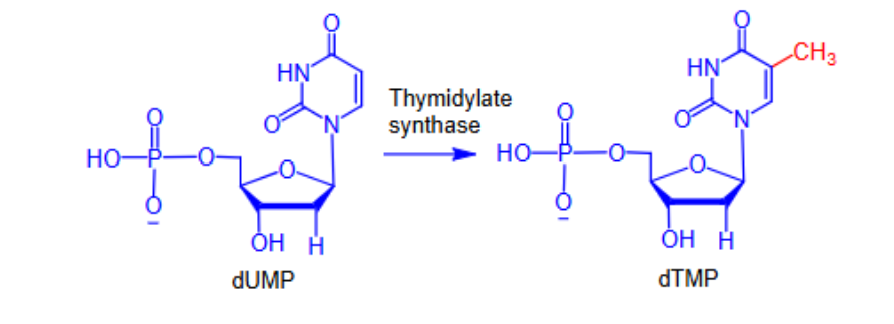
- Pyrimidine antagonists —> inhibit the biosynthesis of dTMP
- Purine antagonists —> inhibit the biosynthesis of AMP and GMP
- Active in the S phase of the cell cycle
Pyrimidine antagonists
- Inhibit the biosynthesis of dTMP
- Inhibit thymidylate synthase (TS), which carries out the rate-limiting step in dTMP synthesis
- Without dTMP, DNA synthesis will stop and the cell will die
5-fluorouracil
- Pyrimidine antagonist
- Dual MoAc:
Inhibits thymidylate synthase by acting as a false substrate (modified molecule that enters and disrupts normal metabolic pathways)
5-dUTP can be incorporated into DNA, leading to chain termination
- Prodrug
- Needs to be converted to its active form, 5-F-dUMP
- 5-F-dUMP is recognized by TS, but not 5-FU
- IV formulation is used to manage colorectal, breast, stomach, and pancreatic cancers
- Topical formulation is used to treat actinic or solar keratosis
- Rapidly cleared
- Most of it undergoes metabolism in the liver by dihydropyrimidine dehydrogenase:
Those with a polymorphism of dihydropyrimidine dehydrogenase are at risk of increased exposure to the drug
Floxuridine
- Pyrimidine antagonist
- Converted to 5-F-dUMP in vivo
- Same MoAc as 5-FU
- Used for the palliative management of GI adenocarcinoma metastatic to the liver
- IV —> regional arterial infusion
- Caution in those with hepatic or renal impairment
- Can be removed via dialysis —> 25-50%
- Lower potential for N/V
- Unlike 5-FU, its PK is not impacted by dihydropyrimidine dehydrogenase
Capecitabine
- Pyrimidine antagonist
- Orally active
- Prodrug of 5-FU
- Converted to 5-F-dUMP in vivo
- Used first-line to treat colorectal cancer
- Used also alone or in combination with docetaxel to treat metastatic breast cancer
- Can cause bone marrow suppression, N/V, hand-and-foot syndrome (leakage from distal capillaries, leading to redness, swelling, and tenderness)
- Lethal DDI with warfarin due to CYP2C9 inhibition
- Negative food effect
Purine antagonists
- Inhibit the de novo biosynthesis of purine nucleotides (AMP and GMP)
- The rate-limiting enzyme in the synthesis of these purine nucleotides is glutamine amidophosphoribosyl transferase
- Glutamine amidophosphoribosyl transferase causes amination of the ribose at the C2 position
- Glycine amino ribonucleotide transformylase (GAR transformylase) requires the formyl group-donating 10-formyl THF
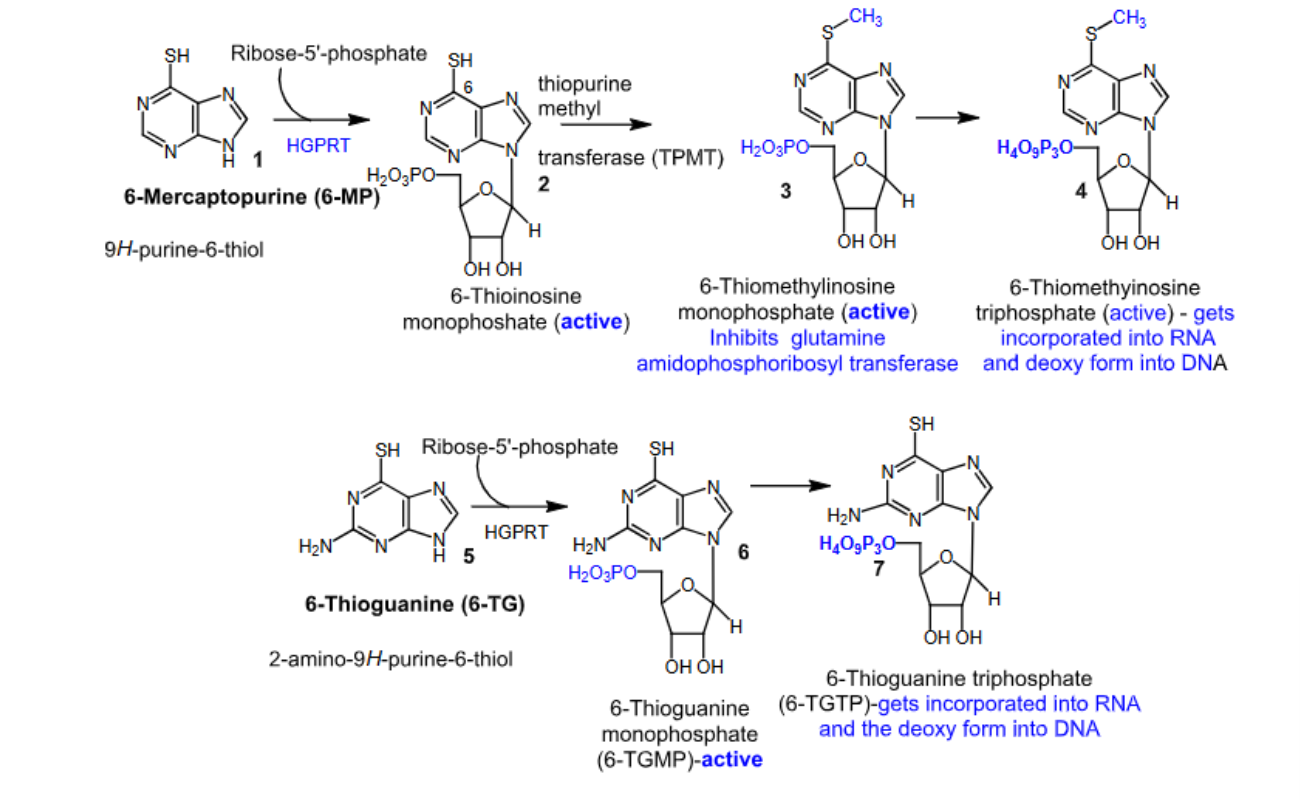
6-mercaptopurine and 6-thioguanine
- Purine antagonists
- Prodrugs
- Converted to their active forms via hypoxanthine guanine phosphoribosyl transferase (HGPRT)
- Inhibit glutamine amidophosphoribosyl transferase, which leads to cell death
- Since 6-mercaptopurine inhibits de novo purine nucleotide biosynthesis rather than incorporation of false nucleotides into DNA, there is a lower risk for mutagenesis and secondary malignancy compared to 6-thioguanine.
- Mechanism of resistance —> deficiency in the activating hypoxanthine guanine phosphoribosyl transferase (HGPRT) enzyme
- Oral
- Negative food effect
- 6-mercaptopurine —> used to treat ALL
- 6-thioguanine —> used to treat AML
- Major metabolizing enzyme is thiopurine S-methyltransferase (TPMT):
Patients with inherited little or no TPMT activity are at an increased risk for severe purinethol toxicity
Poor TPMT metabolizers will have increased levels of active intermediates, and therefore need dose reduction
Cytarabine
- DNA polymerase and chain elongation inhibitor
- Prodrug
- Contains an arabinose instead of a ribose, as seen in cytidine
- Acts as a false substrate for DNA polymerase
- The active form is the triphosphate form, which is incorporated into growing DNA, arresting chain elongation
- Inhibits DNA and RNA polymerases and ribonucleotide reductase (RNA —> DNA) enzymes needed for DNA synthesis
- Active in the S phase of the cell cycle
- Used to treat ANL
- IV
Gemcitabine hydrochloride
- DNA polymerase and chain elongation inhibitor
- Acts as a false substrate for DNA polymerase
- The active form is the triphosphate form, which is incorporated into growing DNA, arresting chain elongation
- The diphosphate form inhibits ribonucleotide reductase
- Longer half-life than cytarabine due to the presence of the gem-difluoromethylene group —> 19 hours
- Used in combination with carboplatin to treat advanced ovarian cancer
- Used first-line in combination with paclitaxel to treat metastatic breast cancer
- Used first-line in combination with cisplatin to treat NSCLC
- Used first-line to treat adenocarcinoma of the pancreas
- IV
Fludarabine phosphate
- DNA polymerase inhibitor
- Inhibits ribonucleotide reductase
- Marketed as the phosphate to improve its aqueous solubility for IV administration
- Triphosphorylated intracellularly by deoxycytidine kinase to the active triphosphate, 2-fluoro-ara-ATP
- Used to treat B-cell CLL
- IV
- Referred to as “AIDS in a bottle” because of its significant immunosuppressive activity
- The C2-F makes it resistant to the degradative action of adenosine deaminase
Antifolates
- Inhibit the function of folic acids
- Inhibit the synthesis of both pyrimidine and purine nucleotides
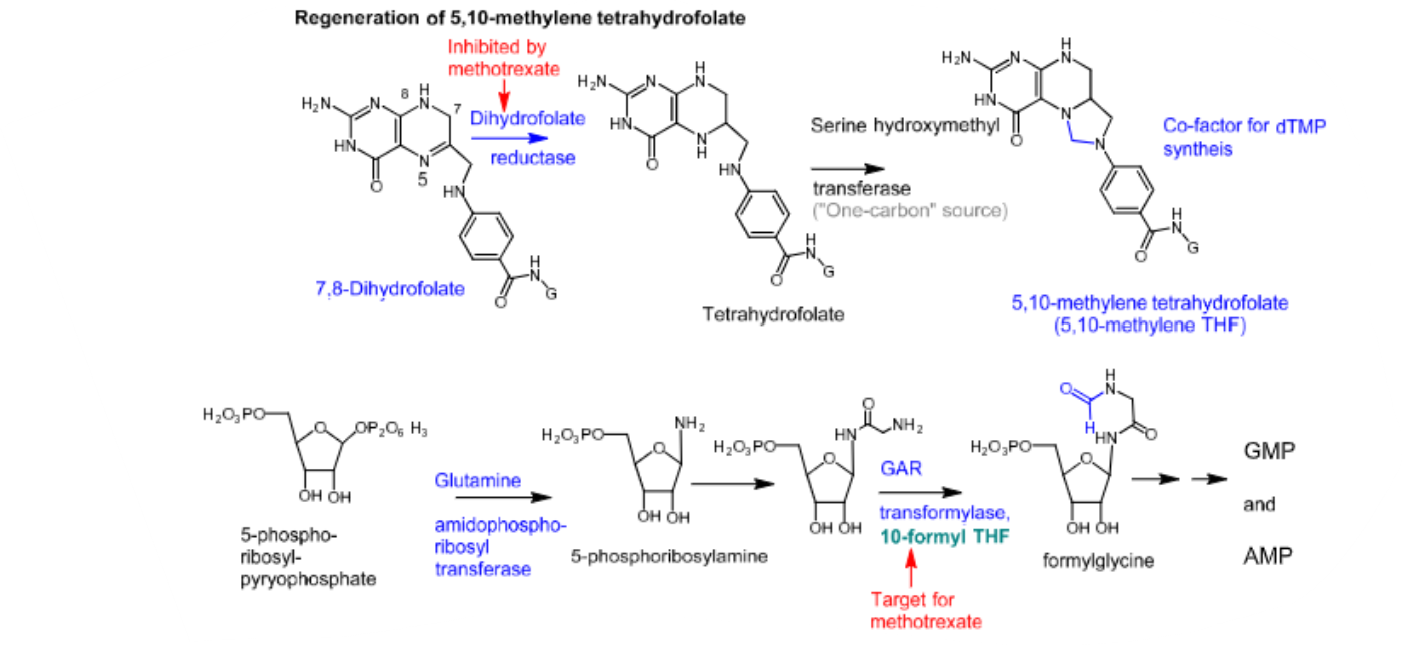
Methotrexate
- Antifolate and antimetabolite
- Inhibits pyrimidine nucleotide synthesis —> via inhibition of DHFR, which ultimately prevents the formation of 5,10-methylene THF
- Inhibits purine nucleotide synthesis —> via inhibition of glycine amide ribonucleotide (GAR) transformylase, which ultimately prevents the formation of GMP and AMP
- Undergoes intracellular folyl polyglutamate synthase (FPGS)-catalyzed polyglutamation, which adds several anionic carboxylate groups to the molecule and traps the drug at the site of action:
Polyglutamation is more effective in tumor cells than in healthy cells, which promotes the selective toxicity of the drug.
- Active during the S phase of the cell cycle
- Used to treat breast, lung, head, and neck cancers
- Used to also treat non-Hodgkin’s lymphoma, psoriasis, and rheumatoid arthritis
- Used off-label to treat MS
- Leucovorin can be used as rescue therapy for methotrexate toxicity —> does not require reduction via DHFR in order to form 5,10-methylene THF
- Cancer cells become resistant to methotrexate over time
- Resistance includes:
Increased DHFR overexpression
Attenuated intracellular polyglutamation
Pemetrexed
- Antifolate and antimetabolite
- Inhibits the synthesis of pyrimidine and purine nucleotides via inhibition of DHFR and GAR transformylase
- Used to treat NSCLC and malignant pleural mesothelioma in combination with cisplatin
- IV
- Can cause bone marrow suppression, neutropenia, nausea, and mucositis
Pentostatin
- Indirect inhibition of ribonucleotide reductase
- Ring-expanded purine
- Inhibits adenosine deaminase —> elevated levels of deoxyadenosine-TP end up inhibiting ribonulceotide reductase
- Used to treat CLL and hairy cell leukemia
- Comparable efficacy to fludarabine, but with a lower risk of toxicity
- Can cause myelosuppression and rashes
Mitosis inhibitors
- Cytotoxic agents
- Disrupt microtubules, which are the basic units of the spindle apparatus:
The spindle apparatus segregates chromosomes between daughter cells during cell division
- During cell division, tubulin undergoes intense, sporadic, and alternating periods of structural growth (polymerization) and erosion (depolymerization) known as “dynamic instability”
- Inhibiting the essential hyperdynamic instability in microtubular structure of the mitotic spindle results in mitotic arrest and apoptosis
- Active in the M phase of the cell cycle
Taxanes
- Commercially synthesized from the more readily available 10-deacetylbaccatin III, which is isolated from a fast growing, renewable ornamental yew, taxus baccata.
- Inhibit the hyperdynamic instability of microtubules by binding to polymerized (elongated) beta-tubulin at a specific hydrophobic receptor site, thus preventing depolymerization.
- Paclitaxel is a natural product, originally isolated from the bark of the slow growing Pacific yew tree, taxus brevifolia.
- Docetaxel and cabazitaxel are semisynthetic
- Due to poor solubility, formulation of taxanes has been an issue:
A mixture of water, alcohol, and Cremophor EL is used. Cremophor EL can cause hypersensitivity, which can be reduced by pretreatment with dexamethasone and antihistamines
Paclitaxel
- Taxane
- Prevents the depolymerization of microtubules
- Used first-line to treat advanced ovarian cancer and NSCLC in combination with cisplatin
- Used to also treat anthracycline-resistant metastatic breast cancer alone or in combination with capecitabine
- Paclitaxel-capecitabine combination therapy is advantageous because paclitaxel upregulates thymidine phosphorylase, which is an enzyme that activates capecitabine
- IV
- DDIs with drugs that are metabolized by CYP2C8
- Major metabolite is less active than the parent
- Resistance arises from cellular efflux by pgp
- Category D teratogen
- Protein bound formulation (Abraxane) —> avoids the hypersensitivity issue, allowing better solubility for IV use, as well as better penetration into tumor cells
Docetaxel
- Taxane
- Prevents the depolymerization of microtubules
- Used in combination with doxorubicin and cyclophosphamide for the adjuvant treatment of patients with operable node-positive breast cancer
- Used to also treat NSCLC, prostate, gastric, and head/neck cancers
- Slightly better solubility than paclitaxel due to the presence of the OH group on C10
- Extensively tissue bound
- Pgp substrate —> cellular efflux by pgp leads to resistance
- Can cause myelosuppression and hypersensitivity reactions (less risk than paclitaxel)
- Category D teratogen
- IV
Cabazitaxel
- Taxane
- Prevents the depolymerization of microtubules
- 7,10-dimethoxy analog of docetaxel —> Lowers its affinity for pgp, leading to better BBB penetration
- Extensively tissue bound
- Less prone to resistance
- Metabolized via CYP3A4 to 10-desmethylcarbazitaxel —> all metabolites are active
- Can become docetaxel after metabolism
- Used to treat docetaxel-resistant metastatic prostate cancer
- Can cause myelosuppression and hypersensitivity reactions
- Category D teratogen
- IV
Ixabepilone
- Derived from the mycobacterium sorangium cellulosum
- Prevents microtubule depolymerization
- Semisynthetic epothilone B analog
- Comparable anticancer activity to epothilone, but has better water solubility and stability because of the lactam
- 2x more potent than paclitaxel in inhibiting microtubule depolymerization
- Low susceptibility to mechanisms of drug resistance, including pgp-mediated drug resistance
- Used in combination with capecitabine in anthracycline- and/or taxane-resistant advanced or metastatic breast cancer or when anthracycline drugs are CI
- Extensively metabolized by CYP3A4
- Use-limiting reactions like peripheral neuropathy and neutropenia prevent its use
- BBW for those with hepatic dysfunction
- IV
Vinca alkaloids
- Vincristine and vinblastine are natural products isolated from catharanthus roseus
- Vinorelbine is semi-synthetic
- Prevent microtubule polymerization
- Bind to alpha/beta-tubulin, and then attenuate the uptake of GTP, which is essential for polymerization
Vincristine
- Vinca alkaloid
- Prevents microtubule polymerization
- Used to treat ALL, Hodgkin’s and non-Hodgkin’s lymphomas, and neuroblastomas
- Category D teratogen
- Severe vesicant
- Cleared slower than vinblastine and vinorelbine —> longest half-life
- IV
Vinblastine
- Vinca alkaloid
- Used to treat breast cancer, Hodgkin’s lymphomas, non-Hodgkin’s lymphomas, and Kaposi sarcomas
- Category D teratogen
- Severe vesicant
- IV
Vinorelbine
- Vinca alkaloid
- Used to treat NSCLC
- Has OBA, but only an IV formulation is available
- Granulocytopenia limits its dosing
- Category D teratogen
- Severe vesicant
- IV
Hydroxyurea
- Cytotoxic agent
- Inhibits ribonucleotide reductase —> prevents the conversion of RNA into DNA
- Used to treat carcinoma of the head/neck with concomitant radiation therapy and CML
- Excellent OBA
- BBW for carcinogenicity
L-asaparginase and peg-asparaginase
- Cytotoxic agents
- L-asparaginase is an enzyme isolated from E. coli
- L-asparaginase converts L-asparagine into aspartic acid and an ammonium ion
- L-asparaginase works via selective killing of leukemic cells due to depletion of plasma asparagine
- Used as a component of a multi-agent chemotherapeutic regimen to treat ALL
- Can cause anaphylaxis and serious allergic reactions
Bortezomib
- Cytotoxic agent
- Reversible inhibitor of the 26S proteasome in mammalian cells:
The 26S proteasome is a large protein complex that degrades ubiquitinated proteins
- Inhibition of the 26S proteasome prevents proteolysis, leading to cell death
- Used to treat multiple myeloma and mantle cell lymphomas
- Can cause diarrhea, nausea, and peripheral neuropathy
- IV
Olaparib (Lynparza)
- Cytotoxic agent
- PARP (poly ADP ribose polymerase) inhibitor
- Inherited mutations in BRCA1 and BRCA2 account for around 20 to 25% of hereditary breast cancers and 15% of ovarian cancers
- Cancer cells use PARP to repair their DNA and continue tumor growth
- Used to treat advanced BRCA-mutated ovarian cancer and BRCA-mutated and HER2-negative metastatic breast cancer:
Patient restriction:
Based on an approved olaparib companion diagnostic test for gBRCA1m or gBRCA2m in the blood
- Oral
Histone deacetylase inhibitors
- Cytotoxic agents
- Uncoiling around histones is accomplished via histone acetyl transferases (HATs), which acetylate the lysine residues in core histones, leading to a less condensed and more transcriptionally active chromatin
- Histone deacetylase (HDAC) removes the acetyl groups (deacetylate) from the lysine residues, leading to the formation of condensed and transcriptionally silenced chromatin
- HDACIs cause hyperacetylated histones, leading to apoptosis
- Normal cells are less vulnerable to HDACI-induced apoptosis
Belinostat
- HDACI
- Hydroxamic acid derivative
- Used to treat relapsed or refractory peripheral T-cell lymphomas
- Oral
- Can cause CV edema, QT prolongation, and skin rashes
- DDIs
Panobinostat
- HDACI
- Hydroxamic acid derivative
- Used to treat multiple myeloma in combination with bortezomib and dexamethasone in patients who have received at least 2 prior regimens, including bortezomib and an immunomodulatory agent
- Oral
- Can cause CV events, cardiac arrhythmias, GI issues, etc.
- DDIs
Glasdegib
- Not a direct cytotoxic agent
- Hedgehog pathway inhibitor:
The hedgehog pathway plays an important role in embryo and fetus development and in the maintenance and regeneration of adult tissues
This pathway is activated in various cancers
Activation of smoothened (SMO), a transmembrane protein, is crucial in the hedgehog signal transduction pathway
- Inhibits SMO activation
- Used to treat newly diagnosed AML in combination with low-dose cytarabine
- Oral
- BBW for embryo/fetal toxicities
Ivosidenib
- Not a direct cytotoxic agent
- Isocitrate dehyrogenase 1 (IDH1) inhibitor:
Normal IDH1 function —> involved in cellular metabolism during the citric acid cyle
IDH1 converts isocitrate to alpha-ketoglutarate (a-KG), which is an important molecule for DNA and histone methylation
Mutant IDH1 and D-2HG —> D-2HG is an oncometabolite that interferes with the function of a-KG-dependent enzymes
- IDH1 mutations occur in 6.6% of AML patients
- Used to treat AML in patients with a susceptible IDH1 mutation
- Oral
- BBW for differentiation syndrome, which can be fatal if not treated
Vorasidenib
- Inhibits mutant IDH1 and IDH2 enzymes
- Used to treat astrocytoma or oligodendroglioma in patients with an IDH1 or IDH2 mutation
- Oral
- Hepatotoxic
- Can increase serum potassium levels
Tyrosine kinase inhibitors
- Not cytotoxic
- Phosphorylation of proteins via kinases is an important mechanism in the signal transduction and regulation of cellular activity
- TKs transfer a phosphate group from ATP to a tyrosine hydroxy group in a protein
- 2 classes:
Receptor tyrosine kinases (RTKs):
Extracellular domain —> binds a specific ligand
Transmembrane domain
Intracellular catalytic domain —> binds and phosphorylates selected substrates
Ex: EGFR AKA ErbB-1/HER1, VEGFR, HER2 AKA Neu/ErbB-2/CD340, and PDGFR
Non-receptor tyrosine kinases (NRTKs):
Cytoplasmic protein kinases
Ex: Bcr-Abl, Src, Pl3Kδ, MEK, B-raf, CDK, and mTOR
- Type 1 TK inhibitors:
Bind to the active conformation of the kinase
- Type 2 TK inhibitors:
Inhibit the enzyme in its inactive conformation
Deemed to have better selectivity
- Promiscuous TKIs (multikinase inhibitors):
Inhibit several kinase enzymes
Imatinib
- Type 2 Bcr-Abl kinase inhibitor
- NRTKI
- Used to treat Ph+ CML, Ph+ ALL, GI stromal tumors, and myelodysplastic/myeloproliferative diseases
- The aberrant Ph chromosome is viewed as the single cause of >90% of adult CML
- Has the greatest effect in the initial (chronic) phase of CML and loses it effectiveness in the accelerated/highly fatal blastic phases
- Mechanisms of resistance:
Point mutations of the Abl kinase domain
Bcr-Abl gene amplification
Pgp-overexpression
Underexpression of OCT1
Activation of Src kinases
- Oral
- OBA of 98%
- PPB of 95%
- Half-life of 18 hours
Nilotinib
- Type 2 Bcr-Abl kinase inhibitor
- NRTKI
- Used to treat Ph+ CML in the chronic phase
- Oral
- Can cause QT prolongation, and this risk is especially pronounced when co-administered with CYP3A4 inhibitors:
QT prolongation is not Bcr-Abl mediated
Dasatinib
- Mixed type 1 and type 2 Bcr-Abl kinase inhibitor
- NRTKI
- Has significant affinity for cellular Src kinases
- Used to treat Ph+ CML and Ph+ ALL
- Oral
- OBA is low due to poor absorption and rapid first-pass CYP3A4-mediated metabolism
- High Vd and highly protein bound
Ruxolitinib (Jakafi)
- Janus kinase (JAK) inhibitor:
JAK is a family of intracellular NRTKs that transduce cytokine-mediated signals via the JAK-STAT pathway
- Inhibits JAK1 and JAK2
- Used to treat intermediate or high-risk myelofibrosis
- Used to also treat polycythemia vera with an inadequate respone to or intolerance to hydroxyurea
- Oral
- Can cause hematological toxicities, infections, and skin cancer
Ibrutinib (Imbruvica)
- Bruton tyrosine kinase (BTK) inhibitor:
BTK is a cytoplasmic NRTK that plays an important role in B cell maturation
- Defective expression of BTK is noted in lymphocytic leukemias
- Irreversible inhibitor of BTK —> binds covalently to Cys481 at the ATP binding site of BTK
- Used to treat CLL
- Oral
- Can cause hematological effects and skin cancer
Acalabrutinib
- Irreversible second-generation BTK inhibitor
- Used to treat CLL and previously treated mantle cell lymphomas
- Oral
- Can cause bone marrow suppression, thrombocytopenia, atrial fibrillation, and skin cancer
Idelalisib
- δ-phosphatidylinositol 3-kinase (Pl3Kδ) inhibitor
- NRTKI
- Pl3Kδ inhibition leads to apoptosis of malignant tumor cells
- Also inhibits chemokine signaling pathways, CXCR4 and CXCR5, which may play a role in CLL
- Used to treat CLL (in combination with rituximab), follicular B-cell-non-Hodgkin lymphomas (after at least 2 prior systemic therapies), and SLL (after at least 2 prior systemic therapies)
- Oral
Duvelisib
- Pl3Kδ/γ inhibitor
- Used to treat CLL
Trametinib
- Mitogen-activated extracellular kinase (MEK) inhibitor:
MEK is a dual threonine and tyrosine kinase that activates mitogen activated protein kinase (MAPK)
- Inhibits MEK1 and MEK2
- Used to treat unresectable or metastatic melanoma in patients with a BRAF V600E or BRAF V600K mutation either as a single agent or in combination with dabrafenib
- Oral
- Can cause dermatological toxicities, hypertension, cardiomyopathy, and pneumonitis
Cobimetinib
- MEK inhibitor
- Inhibits MEK1 and MEK2
- Used to treat unresectable or metastatic melanoma in patients with a BRAF V600E or V600K mutation in combination with vemurafenib (B-Raf inhibitor)
- Oral
- Can cause cutaneous malignancies, hemorrhages, cardiomyopathy, and dermatological toxicities
Dabrafenib
- B-Raf inhibitor
- B-Raf is an NRTK
- BRAF V600 mutations result in constitutive activation of the BRAF pathway
- Inhibitor of certain mutated forms of B-Raf kinases
- Used to treat unresectable or metastatic melanoma in patients with a BRAF V600E as a single agent or in patients with BRAF V600E or BRAF and V600K mutations in combination with trametinib:
Need to confirm BRAF V600E or BRAF V600K mutation status with an approved test prior to treatment
- Oral
- Can cause alopecia, hyperkeratosis, arthralgias, etc.
Sorafenib
- B-raf inhibitor
- Inhibits multiple intracellular kinases:
CRAF, BRAF, and mutant BRAF
- Used to treat advanced renal cell carcinoma and unresectable hepatocellular carcinoma
- Oral
- Can cause skin rashes, hand-foot skin reactions, diarrhea, and hypertension
The cell cycle and checkpoint control
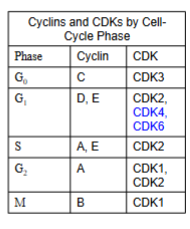
- The most important core cell cycle regulators are known as cyclins —> its enzymes are known as cyclin-dependent kinases (CDKs)
- The cyclin dependent kinase complex is formed by the association of an inactive catalytic unit of a protein kinase (CDK) with the regulatory subunit (cyclin) —> the activated CDK-cyclin complex phosphorylates targeted proteins
CDK inhibitors
- Non-receptor threnonine-serine kinases
- Activate RB
- CDKs are only active when they combine with cyclins to make CDK-cyclin complexes
- The retinoblastoma (RB) protein is a tumor suppressor protein that is dysfunctional in several cancers because RB gets phosphorylated by CDK —> phosphorylated RB does not suppress tumor formation
- RB restricts the cell’s ability to replicate DNA by preventing cell cycle progression from the G1 phase to the S phase:
RB binds and inhibits E2 promoter-binding-protein-dimerization partner (E2F-DP) dimers, which are transcription factors of the E2F family that push the cell into the S phase
RB + E2F-DP —> RB-E2F-DP (cell cycle inhibited)
- Phosphorylation of RB promotes movement from the G1 phase to the S phase
- In normal cell division, when it is time for a cell to enter the S phase, CDK-cyclin complexes phosphorylate RB to pRB, allowing E2F-DP to dissociate from pRB and become active
- Excessive CDK-cyclin phosphorylation of RB —> uncontrolled cell division
Palbociclib (Ibrance)
- CDK inhibitor
- Selectively inhibits CDK4 and CDK6
- Arrests cell cycle progression from the G1 phase to the S phase
- Used to treat HR+ and HER-2-negative advanced or metastatic breast cancer in combination with fulvestrant in women with disease progression following endocrine therapy
- Oral
- Can cause bone marrow suppression and infection
Abemaciclib
- CDK inhibitor
- Selectively inhibits CDK4 and CDK6
- Arrests cell cycle progression from the G1 phase to the S phase
- Used as monotherapy or in combination with fulvestrant to treat HR+ and HER-2-negative advanced or metastatic breast cancer in women with disease progression following endocrine therapy
- Oral
- Can cause GI toxicity, bone marrow suppression, and hepatotoxicity
mTOR kinase inhibitors
- mTOR is a dual serine-threonine kinase:
Activated by forming complexes with other target proteins
- Rapamycin —> natural product isolated from soil bacteria and is sold under the generic name sirolimus and is used as an immunosuppressant
- Temsirolimus and everolimus —> 2 synthetic derivatives of rapamycin that are marketed as anticancer agents
Temsirolimus
- mTOR kinase inhibitor
- Used to treat advanced renal cell carcinoma
- In renal cell carcinoma, mTOR inhibition can exhibit anti-angiogenesis activity
- IV
- Active metabolite is sirolimus (rapamycin), which is a natural product
- Can cause angioedema, bone marrow suppression, and infection
- High Vd
- Metabolized via CYP3A4 to sirolimus (rapamycin) —> an active metabolite
Everolimus
- mTOR inhibitor
- Has antiproliferative and antiangiogenic properties
- Used to treat advanced breast cancer and neuroendocrine tumors
- Used prophylactically to prevent rejection in kidney and liver transplantations
- Oral
- Can cause angioedema, bone marrow suppression, infections, etc.
- OBA of 30%
- High Vd
- Metabolized via CYP3A4 to multiple active metabolites that are weaker than the parent drug
RTKs
- EGFR (AKA ErbB1 or HER1) and EGFR2 (AKA HER2/Neu, ErbB2, and CD340) are closely related membrane-bound TKs:
EGFR over-expression is correlated with decreased life expectancy in solid tumors
HER2 over-expression is a classic feature of treatment-resistant breast, ovarian, lung, and gastric cancers and it endows tumors with what’s been called an “antiapoptotic shield”