test 2- chromosal breaks and translocation mapping methods
1/25
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
26 Terms
how much of genome is non-coding
98%
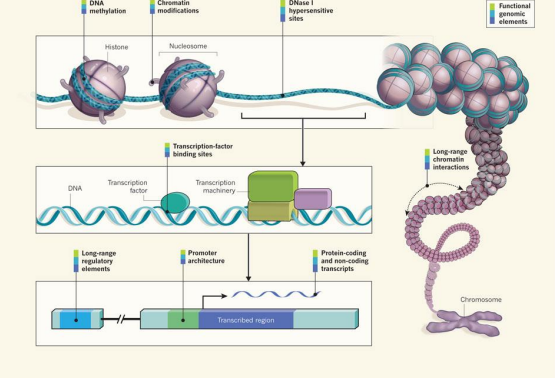
functional element
used to denote a discrete region of the genome that encodes a defined product (e.g. protein) or a reproducible biochemical signature, such as transcription or a specific chromatin structure
beyond the sequence
functional genomic elements that orchestrate the development and function of a human
degree of DNA methylation
chemical modifications to histones
long-range chromatin interactions (ex looping)
alter relative proximities of different chromosomal regions in 3 dimensions
affect transcription
binding activity of transcription-factor proteins
architecture (location & sequence) - of gene regulatory DNA element
promoter region upstream of the point at which transcription of an RNA molecule begins
more distant (long-range) regulatory element
accessibility of the genome to the DNA-cleavage protein DNase I
DNase I hyper sensitive sites
indicate specific sequences at which the binding of transcription factors and transcription machinery proteins has caused nucleosome displacement
catalogs the sequences and quantities of RNA transcripts
non-coding regions
protein-coding regions
ENCODE cell lines (encylopedia of DNA elements)
tiers 1 and 2 widely used cell lines prioritized
tier 1 - highest priority. 3 widely studied cell lines:
K562 erythroleukaemia cells
GM12878, a B-lymphoblastoid cell line
H1 embryonic stem cell line
tier 2 - the secondary priority. Include:
HeLa-S3 cervical carcinoma cells
HepG2 hepatonlastoma cells
primary (non-transformed) human umbilical vein endothelial cells
tier 3 all other ENCODE cell types
ENCODE findings
large amount of human genome 80.4%, is covered by at least one ENCODE-identified element
different RNA types, covering 62% of the genome (although the majority is inside of introns or near genes)
regions highly enriched for histone modifications (56.1%)
excluding RNA elements and broad histone elements, 44.2% of the genome is covered
regions of open chromatin (15.2%)
sites of transcription factor binding (8.1%)
with 19.4% covered by at least one DHS or transcription factor ChiP-seq peak across all cell lines
8.5% of bases are covered by either
a transcription factor binding site motif (4.6%)
or a DHS footprint (5.7%)
could ENCODE improve?
encode project did not assay all cell types, or all transcription factors
it sampled few specialized or developmentally restricted cell lineages
are these proportions underestimatres of the total amount of functional bases?
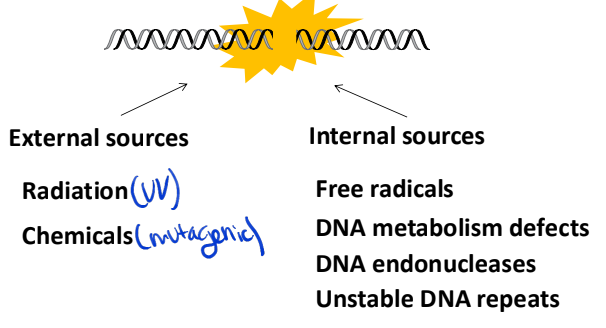
sources of DNA damage
External sources: radiation, chemicals
interal sources: free radicals, DNA metabolism defects, DNA endonucleases, unstable DNA repeats
DNA integrity is essential! Very fragile to damage
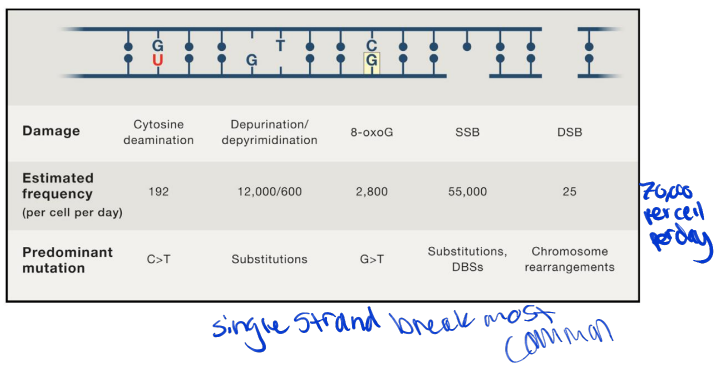
estimated frequencies of DNA lesions and mutations associated with dysfunctional DNA repair
single strand breaks most common
most repaired efficiently ( if not then ds break and they are very dangerous)
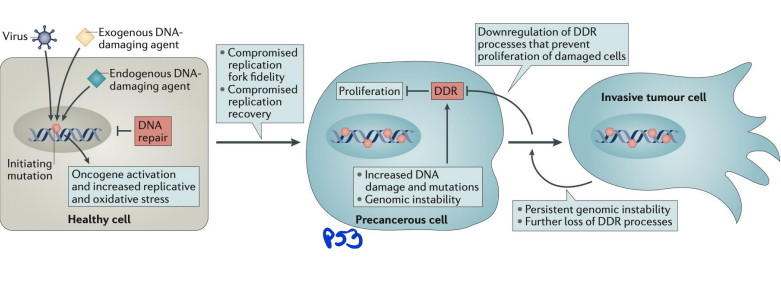
DNA damage and cancers
linked to oncogensis
DNA damage can lead to mutations that drive cancer by activating oncogenes or inactivating tumor suppressor genes. Common sources include UV radiation, chemicals, and oxidative stress. Cells rely on repair mechanisms like mismatch repair (MMR) and homologous recombination (HR) to fix damage, but defects in these pathways (e.g., BRCA1/2 mutations) increase cancer risk
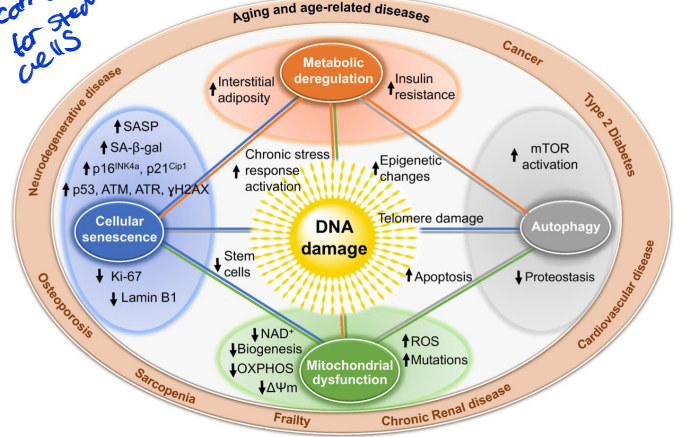
DNA damage and aging
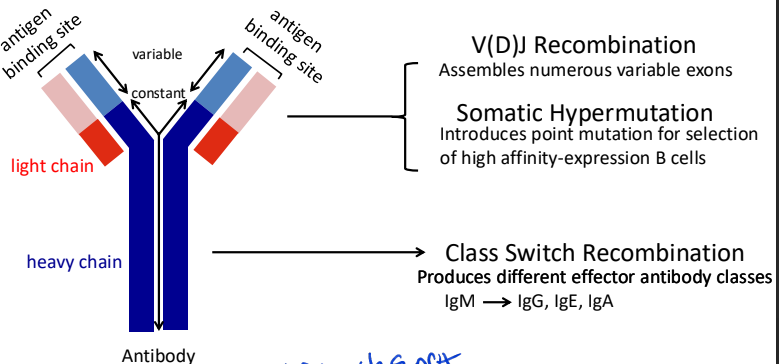
programmed DNA lesions in the immune system
not all DNA breaks are bad some are programmed
major processes that diversify antibody repertoire
V(D)J Recombination – Occurs in developing B and T cells to generate diverse antigen receptors. Mediated by the RAG1/2 enzymes, which introduce double-strand breaks (DSBs) in DNA. Assembles numerous variable exons
Class Switch Recombination (CSR) – Allows B cells to switch antibody isotypes (e.g., IgM to IgG) by creating targeted DSBs in immunoglobulin genes, facilitated by Activation-Induced Cytidine Deaminase (AID).
Somatic Hypermutation (SHM) – Introduces point mutations in immunoglobulin genes to enhance antibody affinity, also driven by AID.
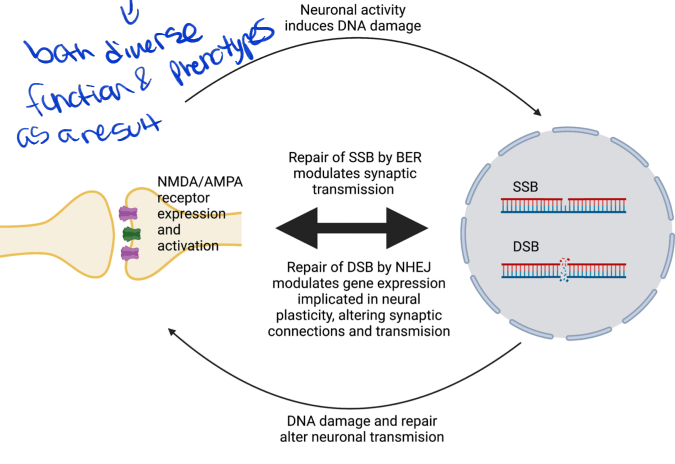
programmed dna damage in neural plasticity
both diverse function and phenotypes as a result
ChiP-Seq (DSB detection via locating chromatin-bound DSB repair factors)
pulls down proteins specifically binding to broken ends or processed ends
dna repair factors recruited to dsbreak location
not precise or specific need to couple w/ other method
BLESS (genome-wide mapping of DNA ends)
a benchmark DSB detection method
direct in situ breakls labeling, enrichment on streptavidin and next-generation sequencing
nucleotide resolution - determine position of event
Key Steps in BLESS:
DSB Labeling – Biotinylated oligonucleotides bind exposed DNA ends in fixed cells.
Enrichment – Streptavidin purification isolates labeled DNA fragments.
Sequencing – High-throughput sequencing identifies DSB locations across the genome.
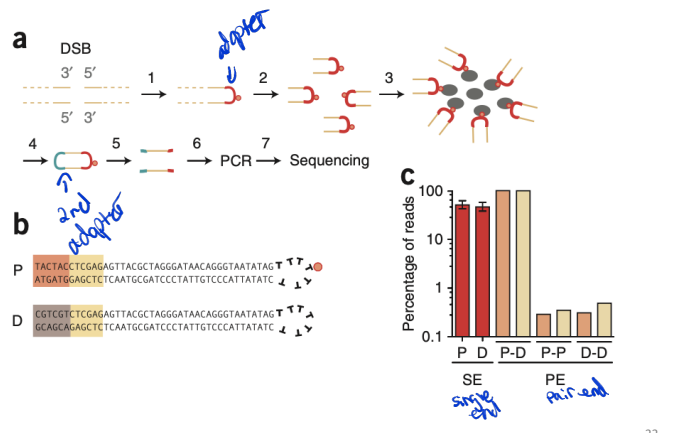
BLESS limitations
the possibility of creating fixation-induced DNA breaks
high background signals
inefficient ligation of the linkers to the DNA ends
it is occasionally unable to detect both ends of the same DSB
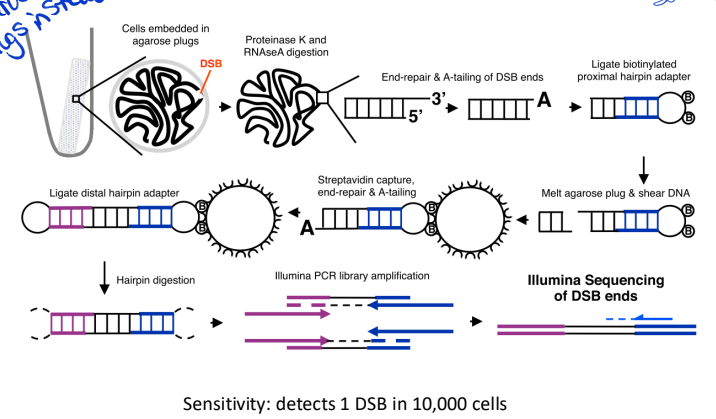
End-seq (genome-wide mapping of DNA ends)
cells are embedded in agarose plugs to avoid fixation with formaldehyde
more widely used - better specificty and sensitivity
no formaldehyde treatment agarose plugs instead
Key Steps in END-seq:
DSB End Processing – DNA breaks are blunted and labeled with a biotinylated adapter.
Purification & Enrichment – Biotin-tagged DNA is isolated using streptavidin beads.
Library Preparation & Sequencing – Labeled DNA ends are sequenced to determine break sites.
end-seq vs bless
less background and better sensitivity
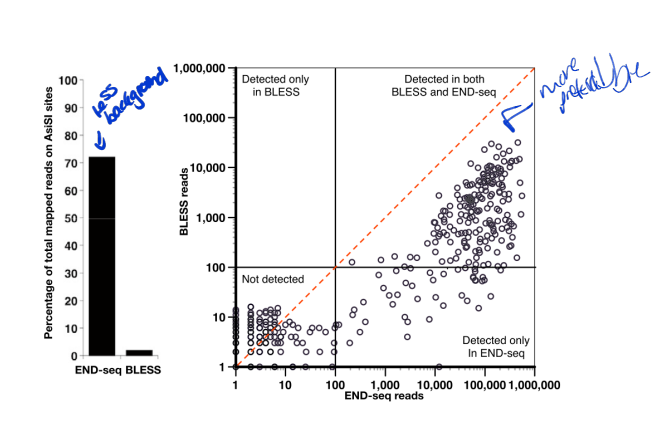
end-seq pro and con
pros: high sensitivity; provide snapshots of unrepaired DSBs
cons: require efficient tagging of DSBs; may induce DSBs by cell manipulation
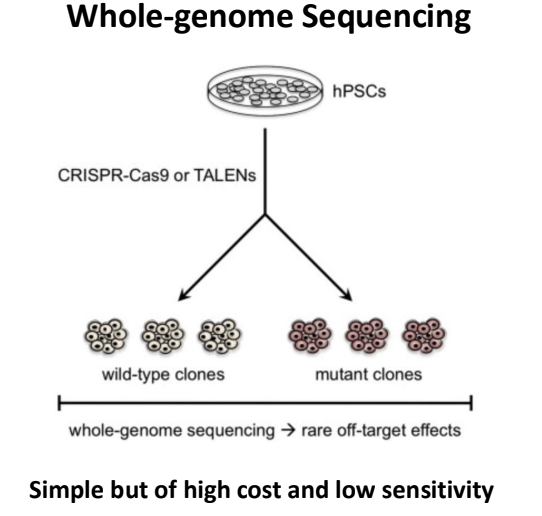
whole-genome sequencing (genome-wide mapping of repaired DNA DSBs)
if DSB- will have mutation signatures
most will be junk sequences so you need to go more specific
guide-seq (genome-wide mapping of repaired DNA DSBs)
GUIDE-Seq consists of two stages. In stage I, blunt-ended DSBs in the genomes of living human cells are tagged by integration of a blunt, double-stranded oligodeoxynucleotide (dsODN) at these breaks by means of an end-joining process consistent with NHEJ. In stage II, dsODN integration sites in genomice DNA are precisely mapped at the nucleotide level using unbiased amplification and next-generation sequencing
enriched through PCR so highly sensitive
only blunt-end breaks
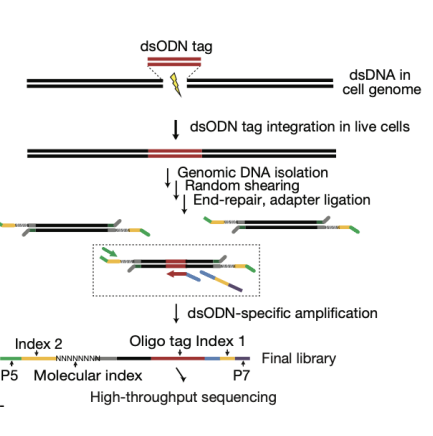
guide-seq pros and cons
pros: unbiased, sensitive
cons: currently limited use for blunt-ended DSBs
LAM-HTGTS (linear amplification-mediated high-throughput genome-wide translocation sequencing) (genome-wide mapping of repaired DNA DSBs)
a bait DSB generated (ectopically or endogenously) at a known genomic site serves as a translocation site for other prey DSBs generated in the genome. A step of linear amplification PCR is employed to enrich bait break associated translocations
highest sensitivity so far (bait reason)
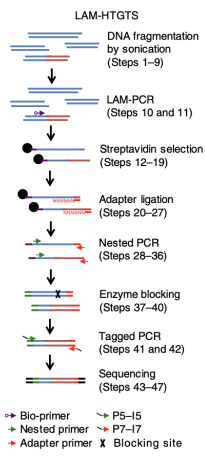
LAM-HTGTS pros and cons
pros: highly sensitive to bait-break associated translocations, low background
cons: require a known bait to break to detect other breaks (have to creat break and then see what breaks are around), can only detect breaks translocated to the bait, has lower efficiency in detecting DSBs generated in regions that are not topologically associated with the bait DSB. Also more efficient in cis than trans for bait
S1-END-seq (DNA single-strand break detection methods)
it utilizes S1 single strand DNA endonuclease to convert ssDNA regions to DSBs, whcih are then detected via End-Seq
many more ssDNA than dsDNA
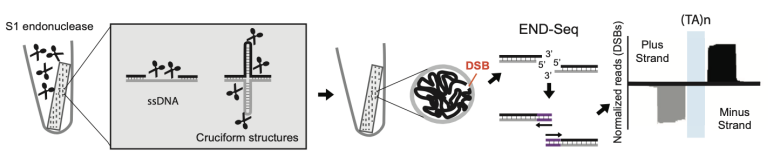
detection of DNA secondary structures via S1-END-seq
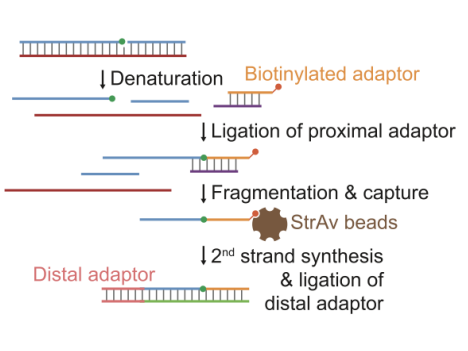
GLOE-Seq (genome-wide ligation of 3’-OH ends followed by sequencing)(DNA single-strand break detection methods)
It detects free 3’-OH termini resulted from SSBs, lesions or other repair intermediates. In GLOE-Seq, genomic DNA is heat-denatured and ligated to a biotinylated adapter with the assistane of a splinter oligonucleotide, followed by fragmentation and capture on streptavidin beads