5-Hemoglobinopatías
1/17
Earn XP
Description and Tags
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
18 Terms
Hemoglobinas normales que aparecen en el ADULTO y patológicas
Hemoglobinas normales en ADULTO:
Hemoglobinas HbA:
2 alfa
2 beta
Hemoglobina HbA2:
2 alfa
2 delta
Hemoglobina fetal HbF (1% en adulto, 80% al nacimiento):
2 alfa
2 gamma
Hemoglobina PATOLÓGICA:
Hemoglobina S HbS (Cadena BETA: cambio de AA glutamico en posición 6 hay VALINA)
2 alfas
2 betas (truchas ambas o solo 1)
Puede dar: CRISIS VASO OCLUSIVAS (x anemia falciforme, cambia la forma del eritrocito)
Desarrollo
Hemoglobina del: EMBRIÓN
EMBRIÓN: Primeras 8 semanas
Genes: Z (cromosoma 16) y epsilon (Cromosoma 11) = Hb DE GOWER
Genes: Z (cromosoma 16) y gamma/beta (Cromosoma 11) = Hb DE PORTLAND
Desarrollo
Hemoglobina del: FETO
FETO: Luego de las primeras 8 semanas
Hemoglobina fetal HbF (llega al 90%, disminuye un 50-80% luego del nacimiento, hasta el 6to mes 5% y en el adulto es de 1%):
2 alfa
2 gamma
Un gen la inhibe luego del nacimiento
Porcentajes en el adulto de los distintos tipos de Hb
HbA= 97% (Alfa y Beta) (beta con glucosa= Hb glicosilada)
HbA2= 2% (Alfa y Delta)
HbF= 1% (Alfa y Gamma)
Hemoglobina Glicosilada
Se une la glucosa a la hemoglobina hasta un: 5,7-6%
Rangos:
⬆5,7% de Hb Glicosilada= HIPERglicemias en últimos 3 meses previos❌
⬇5,7% de Hemoglobina glicosilado = SANO/ NORMAL✅
Últimos 3 meses, porque el glóbulo rojo vive 120 días = 3 meses aprox
Desarrollo
Hemoglobina del: RECIÉN NACIDO
Recién nacido:
HbF = 50 - 80% (Alfa y Gamma)
Hb de GOWER ( Z y epsilon)
Hb de PORTLAND ( Zeta y gamma o beta)
A los 6 meses:
HbF= 5% (Alfa y Gamma)
Ejecicio: ¿Que presenta el paciente ADULTO?
HbS = 48%
HbA = 50%
HbA2 = 2%
HbF= 1
PRESENCIA HbS= Anemia falciforme hemolítica (¿pero es portador o enfermo?)
Produce HbA = PORTADOR
Una de sus cadenas BETA (con valina) esta alterada y otra no, porque puede formar HbA
HETEROCIGOTO
Minima anemia o no tiene anemia.
Recordar:
HbA (Alfa y Beta)= 95% es lo normal.
Anemia falciforme: Forma HbS, es PATOLOGICA.
Altera la cadena BETA: cambio de AA glutámico en posición 6 x VALINA
Ejecicio: ¿Que presenta el paciente ADULTO?
HbS = 90%
HbA = 0%
HbF= 10%
PRESENCIA HbS= Anemia falciforme hemolítica (¿pero es portador o enfermo?)
NO Produce HbA = ENFERMO
AMBAS cadenas BETA estan alteradas, NO puede formar HbA
HOMOCIGOTO
En cambio forma HbF para compensar la falta HbA
Recordar:
HbA (Alfa y Beta) = 95% es lo normal.
Anemia falciforme: Forma HbS, es PATOLOGICA.
Altera la cadena BETA: cambio de AA glutámico en posición 6 x VALINA
Ejecicio: ¿Que presenta el paciente ADULTO?
HbA = 85%
HbF= 10%
HbA2 = 5%
Falta un gen Beta: ANEMIA MICROCÍTICA
⬇HbA (normal es 95%)
Compensa con HbF = ⬆HbF
HbA2 normal = 2% (no quiere trabajar)
PORTADOR: heterocigoto ⬆⬆HbA2
TALASEMIA MAYOR= homocigoto ⬆⬆HbF (NO hay HbA)
Anemia Drepanocítica (SCD) Sickle cell disasse
Herencia homocigota de genes HbS
Ambos genes forman cadenas BETA: con VALINA
Drepanocitos
Células con forma de Hoz
Rígidos
Se rompen fácilmente
Hb con O2 = parece un glóbulo rojo normal con forma BICONCAVA
Hb SIN O2 = forma de drepanocitos forma de hoz
Crisis vaso oclusivas
Oclusión vascular (COV)
Isquemia, Dolor, Edema, lesiones
Hemograma (ver los drepanocitos)
Electroforesis de Hb (ver HbS)
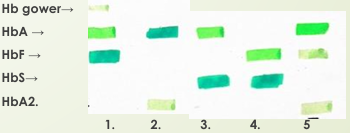
Ejercicio
1) Recién nacido (presencia Hb Gower) Alto HbF (80%) y algo de HbA
2) Adulto Normal: HbA (97%) y algo de HbA2 (2%)
3) CON HbS. Tiene HbA = PORTADOR
4) CON HbS. NO tiene HbA= ENFERMO (HbF para compensar)
5) B-Talasemia. ⬇⬇ HbA (menor a 97%), algo de HbA2 (2%) y HbF (1%)
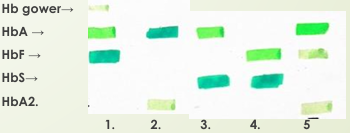
Comparación de hemoglobinas
Recien nacido
⬆⬆HbF (80%), ⬇HbA y Hb gower
Adulto
⬆⬆HbA (97%), algo de HbA2 (2%)
Portador falciforme
HbS y con HbA
Falciforme severa
⬆⬆HbS y ⬆HbF (SIN HbA)
B-Talasemia
⬇⬇HbA , con HbA2 y ⬆HbF
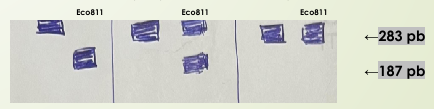
¿Que corta la enzima restrictiva (Eco 81L)? ¿Como es en un paciente con anemia falciforme y un portador?
Corta al ADN sano, quedando menor tamaño del ADN
Anemia falciforme. Homocigoto (ambas copias están mal)= No corta nada
Portador anemia falciforme. Heterocigoto (una de las copias esta mal)= Corta una no todo

Beta Talasemia
Beta Talasemias: problema con cadena beta
2 genes,
1 gen beta de la madre
1 gen beta del padre
Presentes en cromosoma 11
En el nacimiento, si falta beta: le da Anemia x ⬇HbF (Alfa y Gamma)
A la hora de producir ❌HbA (Alfa y Beta), no puede porque es una talasemia y no tienen beta, dando como resultado una anemia.
Menor= 1 solo gen afectado, Anemia microcitica leve (⬆⬆ HbA2)
Mayor= 2 genes afectados, Anemia severa esplenomegalia (⬆⬆HbF)
HIPERreactividad de la medula ósea afecta los huesos
Alfa talasemia
Alfa Talasemias: problema con cadena alfa
4 genes=
2 copias de Alfa 1 (1 madre y otro padre)
2 copias de Alfa 2 (1 madre y otro del padre)
Ambos del cromosoma 16
A/A : A/- = Portadores asintomáticos (1 gen afectado)
A/- : A/- = Anemia microcítica leve (2 genes afectados)
A/- : -/- = Anemia hemolítica sintomática y esplenomegalia
-/- : -/- = Incompatible con la vida
Significado de las siglas: SCD, COV, ECF, TDT
Siglas de las enfermedades:
SCD= Sickle cell disase, Anemia drepanocítica
COV= Crisis vaso oclusivas
ECF= Enfermedad de células Falciformes (Anemia drepanocítica severa)
TDT= Talasemia dependiente de transfusiones (Beta talasemia)
Accion de las CRISPR-Cas9
CRISPR-Cas9 es una enzima (editora)
CTX 001= pacientes con Anemia drepanocítica y Beta talasemia Severas
Modifica las células madre: (cromosoma 2, elimina potenciador)
Elimina el gen represor BCL11A inhibidor de ❌HbF = ⬆⬆Expresión HbF
✅HbF se comporta como una HbA, solucionando la talasemia.
Hemoglobina M
Mutación de HISTIDINA x TIROSINA (en cadena alfa o en beta)
SIN ❌❌HISTIDINA PROXIMAL
Tirosina ↔ Fe3+
Histidina ↔ Fe2+
Tiene: ➕Afinidad (le cuesta soltar más a el oxigeno) = HIPOXIA, ⬆Eritropoyetina, Poliglobulia
Mutación en ALFA: Altera equilibrio R-T (NO sensible al efecto de H+, efecto Bohr)
Mutación en BETA: SI es sensible al efecto Bohr