PATHO Notes – Hematologic Disorders: RBC Disorders 🧬 – IRAT 8 (Part 1-Primary Homeostasis )
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
88 Terms
What types of bleeding disorders typically cause bruising and frequent nosebleeds?
Platelet or von Willebrand factor (vWF) defects causing mucocutaneous bleeding.
What is the most likely diagnosis for a child with easy bruising, epistaxis, and a maternal history of bleeding?
Von Willebrand Disease (vWD).
What are other possible diagnoses that could explain this child’s bleeding symptoms?
Platelet function disorders, mild hemophilia, or Factor XI deficiency.
What laboratory studies should be ordered to evaluate this child’s bleeding tendency?
CBC, PT, aPTT, Bleeding time or PFA, vWF antigen, Ristocetin test, and Factor VIII level.
What laboratory findings are typical in von Willebrand disease?
Normal platelets and PT
prolonged aPTT and bleeding time
decreased vWF and ristocetin activity.
Why did this child not bleed excessively during circumcision?
The disease is mild and newborns have higher vWF levels that temporarily mask symptoms.
What is the purpose of hemostasis?
To repair blood vessel damage by forming a thrombus to stop bleeding.
What are the two main stages of hemostasis?
1⃣ Primary hemostasis and 2⃣ Secondary hemostasis.
What happens during primary hemostasis?
A weak platelet plug forms via interaction between platelets and the vessel wall.
What happens during secondary hemostasis?
The platelet plug is stabilized through the coagulation cascade.
What organ produces most coagulation factors?
The liver.
What enzyme activates vitamin K in the liver?
Epoxide reductase.
Which clotting factors are vitamin K–dependent?
Factors II, VII, IX, and X.
What happens in vitamin K deficiency?
Impaired synthesis of clotting factors II, VII, IX, and X → prolonged bleeding.
How does liver disease affect coagulation?
Decreased production of clotting factors → bleeding tendency.
Which hemophilias affect the intrinsic pathway?
Hemophilia A → Factor VIII deficiency (7)
Hemophilia B → Factor IX deficiency (9)
Hemophilia C → Factor XI deficiency (11)
Which pathway is measured by PT and which by aPTT?
PT → Extrinsic pathway (Factor VII)
aPTT → Intrinsic pathway (Factors XII, XI, IX, VIII)
🩸 Primary Hemostasis Flashcards (Q & A Format)
Questions
What is the main goal of primary hemostasis?
What is Step 1 of primary hemostasis?
What is Step 2 of primary hemostasis?
What does von Willebrand factor (vWF) bind to during platelet adhesion?
What receptor on platelets binds to vWF?
What is Step 3 of primary hemostasis?
What does ADP released from platelet granules do?
What does thromboxane A₂ (TXA₂) synthesized by platelet COX promote?
What is Step 4 of primary hemostasis?
How do platelets aggregate during Step 4?
What process is needed to stabilize the weak platelet plug?
Answers
To form a weak platelet plug at the site of vessel injury.
Transient vasoconstriction of the damaged vessel to reduce blood flow.
Platelet adhesion to the exposed surface of the disrupted vessel.
Exposed collagen on the damaged vessel wall.
The GPIb receptor.
Platelet degranulation.
Promotes expression of GPIIb/IIIa receptors on platelets.
Promotes platelet aggregation and vasoconstriction.
Platelet aggregation at the injury site.
GPIIb/IIIa receptors on platelets bind fibrinogen, linking platelets together.
The coagulation cascade (secondary hemostasis).
What are the 4 Primary Hemostasis Disorders?
Immune Thrombocytopenia
Microangiopathic Hemolytic Anemia
Bernard-Soulier
Glanzmann Thrombasthenia
What are disorders of primary hemostasis usually due to?
Platelet abnormalities (quantitative or qualitative)
What are the main clinical features of disorders of primary hemostasis?
Mucosal and skin bleeding
What types of mucosal bleeding are seen in platelet disorders?
Epistaxis, hemoptysis, GI bleeding, hematuria, menorrhagia, and severe intracranial bleeding
What types of skin bleeding are seen in platelet disorders?
Petechiae (1–2 mm), purpura (>3 mm), and ecchymoses (>1 cm).
Why are conditions of primary hemostasis primarily platelet disorders?
Because platelet adhesion and aggregation form the initial platelet plug in hemostasis.
What is the normal platelet count range?
150 – 400 K/µL.
At what platelet count do symptoms typically begin to appear?
Below 50 K/µL.
What is the normal bleeding time and when is it prolonged?
Normal: 2–7 minutes; prolonged in platelet disorders.
What does a blood smear evaluate in platelet disorders?
Number and size of platelets.
What is assessed by bone marrow biopsy in platelet disorders?
Presence and number of megakaryocytes (platelet precursors).
What is the most common cause of thrombocytopenia in both children and adults?
Immune Thrombocytopenia (ITP).
What type of condition is ITP?
An autoimmune disorder.
When does acute ITP typically occur and in which population?
In children following a viral infection or immunization.
In whom does chronic ITP usually develop?
Adults, especially women of childbearing age.
What is the pathophysiologic mechanism of ITP?
IgG antibodies target platelet antigens (commonly GPIIb/IIIa), marking them for destruction.
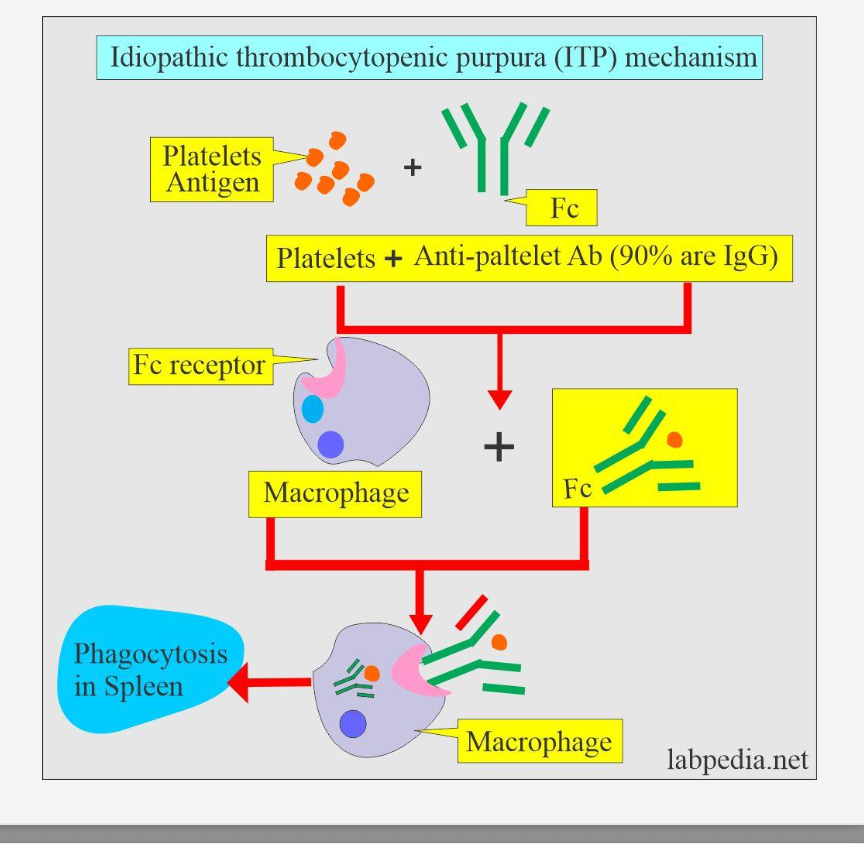
Where are the autoantibodies against platelets produced in ITP?
By plasma cells in the spleen.
How are antibody-coated platelets destroyed in ITP?
They are phagocytosed by splenic macrophages, leading to thrombocytopenia.
Why is the spleen central to ITP pathogenesis?
It is both the site of antibody production and platelet destruction.
How does acute ITP typically resolve?
It is self-limited and resolves spontaneously within weeks.
How can chronic ITP in women affect newborns?
Maternal IgG antibodies cross the placenta, causing temporary thrombocytopenia in the infant.
What type of bleeding pattern do ITP symptoms reflect?
A primary hemostasis disorder (due to platelet dysfunction).
What are the main clinical signs of ITP?
Mucosal bleeding: epistaxis, GI bleeding, menorrhagia
Skin bleeding: petechiae, purpura, ecchymoses
What are the hallmark laboratory findings in ITP?
↓ Platelet count (<50k)
Normal PT and PTT *comes in handy for secondary hemostasis (Coagulation Cascade)
↑ Bleeding time (Prolonged (with platelet disorders)
Bone marrow: ↑ megakaryocytes
Why are megakaryocytes increased in the bone marrow of ITP patients?
The body increases platelet production to compensate for peripheral destruction.
Why is splenomegaly uncommon in ITP?
True ITP rarely causes splenomegaly; its presence suggests another diagnosis (e.g., leukemia or hypersplenism).
What finding should raise suspicion for ITP in children?
Thrombocytopenia and petechiae after a recent viral illness.
A 7-year-old child develops petechiae and mild mucosal bleeding two weeks after recovering from a viral infection. Platelet count is 30,000/µL, PT and PTT are normal, and bone marrow biopsy shows increased megakaryocytes.
Q: What is the likely diagnosis and cause?
A: Acute ITP — IgG antibodies against GPIIb/IIIa cause splenic macrophage destruction of platelets following viral activation.
A 29-year-old woman with heavy menstrual bleeding gives birth to an infant who develops petechiae at 2 days old. Labs show low platelets in both mother and newborn; PT/PTT are normal.
Q: What caused the newborn’s thrombocytopenia?
A: Maternal IgG autoantibodies crossed the placenta, causing transient neonatal thrombocytopenia secondary to maternal chronic ITP.
What does MAHA stand for?
Microangiopathic Hemolytic Anemia.
🧬 Microangiopathic Hemolytic Anemia (MAHA) Flashcards:
What causes MAHA?
Pathologic formation of platelet thrombi in small vessels.
What are the two primary causes of MAHA? **more important to know
Thrombotic Thrombocytopenic Purpura (TTP) and Hemolytic Uremic Syndrome (HUS).
What are secondary causes of MAHA?
SLE, HELLP syndrome, HTN emergency, DIC, and drug-induced hemolytic anemia.
What happens to platelets in MAHA pathophysiology?
What happens to RBCs in MAHA pathophysiology?
Platelets are consumed due to the formation of microthrombi.
RBCs are sheared by the microthrombi, forming schistocytes (Helmet Cells)
What is the cause of TTP?
Decreased ADAMTS13.
What is ADAMTS13 and what is its normal function?
It is an enzyme that normally cleaves vWF multimers to prevent clot formation.
What happens when vWF multimers are not cleaved?
Uncleaved vWF multimers cause abnormal platelet adhesion and microthrombi formation
What is the usual cause of decreased ADAMTS13?
An acquired antibody, most common in adult females.
Hemolytic Uremic Syndrome (HUS) — Etiology (Cause)
Shiga toxin (E. coli O157:H7) → endothelial injury → platelet microthrombi
What infection is most commonly associated with HUS?
E. coli O157:H7 dysentery from undercooked beef—>
How does E. coli cause HUS?
E. coli verotoxin damages the endothelium—> platelet microthrombi formation.
What are the main clinical features of MAHA?
Skin and mucosal bleeding
Fever
Which organ system is most affected in HUS?
The kidneys → renal insufficiency (most common in HUS).
Which organ system is most affected in TTP?
The central nervous system → CNS abnormalities (most common in TTP).
🧪 Labs
Q: What are the key lab findings in MAHA?
A:
Thrombocytopenia (low platelets)
Increased bleeding time
Normal PT/PTT
Anemia with schistocytes
Increased megakaryocytes in bone marrow biopsy
⚖ Comparison: TTP vs HUS
Feature | TTP (Thrombotic Thrombocytopenic Purpura) | HUS (Hemolytic Uremic Syndrome) |
|---|---|---|
Main Cause | ↓ ADAMTS13 enzyme | Endothelial damage by infection or drugs |
Mechanism | Uncleaved vWF multimers → ↑ platelet adhesion → microthrombi | E. coli O157:H7 verotoxin damages endothelium → platelet microthrombi |
Typical Patient | Adult females | Children after diarrheal illness (E. coli) |
Organ System Most Affected | CNS (neurologic abnormalities) | Kidneys (renal insufficiency) |
Peripheral Smear | Schistocytes (fragmented RBCs) | Schistocytes (fragmented RBCs) |
PT/PTT | Normal | Normal |
Bleeding Time | Increased | Increased |
Bone Marrow | ↑ Megakaryocytes | ↑ Megakaryocytes |
🧫 Bernard–Soulier Syndrome Flashcards
What type of hemostatic disorder is Bernard–Soulier syndrome?
A disorder of primary hemostasis.
What is the inheritance pattern of Bernard–Soulier syndrome?
Autosomal recessive.
What is the primary defect in Bernard–Soulier syndrome?
A deficiency of the Gp1b receptor on platelets.
What does Gp1b normally bind to during hemostasis?
Von Willebrand factor (vWF) — necessary for platelet adhesion to the vessel wall.
How does Gp1b deficiency affect platelet function?
It causes impaired platelet adhesion to the vessel wall.
What type of bleeding symptoms occur in Bernard–Soulier syndrome?
Mucosal and skin bleeding (epistaxis and petechiae)
What are the platelet count and size findings in Bernard–Soulier syndrome?
Low to normal platelet count; platelets may appear large on smear.
How is bleeding time affected in Bernard–Soulier syndrome?
Prolonged (characteristic of platelet disorders).
How are PT and PTT affected in Bernard–Soulier syndrome?
Normal, because secondary hemostasis is not affected.
What are the key laboratory findings in Bernard–Soulier syndrome?
Platelet count/size: Low to normal
Bleeding time: Prolonged (platelet disorder)
PT/PTT: Normal (secondary hemostasis unaffected)
🧬 Glanzmann Thrombasthenia – Flashcards
What type of hemostatic disorder is Glanzmann Thrombasthenia?
A: A primary hemostasis disorder.
What is the inheritance pattern of Glanzmann Thrombasthenia?
Autosomal recessive.
What is the primary defect in Glanzmann Thrombasthenia?
What does the GpIIb/IIIa receptor normally bind to?
Deficiency of the GpIIb/IIIa receptor complex on platelets;
Fibrinogen, which cross-links platelets during aggregation.
How does a GpIIb/IIIa deficiency affect platelet function?
Causes impaired platelet aggregation.
What type of bleeding symptoms are seen in Glanzmann Thrombasthenia?
Mucosal and skin bleeding (e.g., epistaxis, petechiae).
What are the key lab findings in Glanzmann Thrombasthenia?
Platelet count: Low to normal
Bleeding time: Prolonged
PT/PTT: Normal (secondary hemostasis unaffected)
What is the main difference between platelet adhesion and aggregation defects?
Adhesion defect: Platelets can’t stick to the vessel wall (e.g., GpIb deficiency – Bernard-Soulier)
Aggregation defect: Platelets can’t stick to each other (e.g., GpIIb/IIIa deficiency – Glanzmann)
Which disorders involve platelet consumption rather than receptor defects?
A: TTP, HUS, and MAHA — all involve microthrombi formation and RBC shearing (schistocytes).
What type of bleeding is common to all primary hemostasis disorders?
A: Mucocutaneous bleeding (epistaxis, petechiae, easy bruising).
🧠 Learning Tricks
🩸 “Bernard can’t stick, Glanzmann can’t clump.”
→ Bernard-Soulier = adhesion problem (GpIb)
→ Glanzmann = aggregation problem (GpIIb/IIIa)🔬 “Bs before Gs” → Adhesion (Bernard-Soulier) occurs before aggregation (Glanzmann) in the hemostasis process.
⚖ “Normal PT/PTT = primary problem.”
→ If PT/PTT are normal but bleeding time is prolonged → it’s platelet-related, not clotting factor-related.
📊 Comparison Table – Platelet Disorders Overview
Feature | Bernard–Soulier Syndrome | Glanzmann Thrombasthenia | Immune Thrombocytopenia (ITP) | TTP / HUS (MAHA) |
|---|---|---|---|---|
Defect Type | Platelet adhesion | Platelet aggregation | Platelet destruction (autoimmune) | Platelet consumption (microthrombi) |
Key Protein/Receptor | ↓ GpIb | ↓ GpIIb/IIIa | Autoantibodies to platelets | ↓ ADAMTS13 (TTP) / Endothelial damage (HUS) |
Pathophysiology | Can’t bind vWF | Can’t bind fibrinogen | Antibody-coated platelets destroyed in spleen | Microthrombi consume platelets & shear RBCs |
Bleeding Type | Mucosal, skin | Mucosal, skin | Mucosal, skin | Mucosal, skin + organ damage |
Platelet Count | ↓ / normal | ↓ / normal | ↓ | ↓ |
Bleeding Time | ↑ | ↑ | ↑ | ↑ |
PT/PTT | Normal | Normal | Normal | Normal |
Bone Marrow | Normal / ↑ megakaryocytes | Normal | ↑ megakaryocytes | ↑ megakaryocytes |
Unique Clues | Giant platelets | No aggregation with ADP or fibrinogen | Follows viral infection (esp. children) | Schistocytes + renal (HUS) or CNS (TTP) findings |
🧠 Study Check 1 – TTP
A 28-year-old female presents with fever, confusion, and skin bleeding.
Blood smear shows schistocytes, and labs reveal thrombocytopenia with normal PT/PTT.
ADAMTS13 activity is severely decreased due to an acquired autoantibody.
Which of the following is the most likely underlying mechanism?
a) Autoimmune destruction of platelets in the spleen
b) Endothelial damage caused by E. coli verotoxin
c) Lack of ADAMTS13 to cleave von Willebrand factor multimers
d) Deficiency of GpIIb/IIIa receptors leading to impaired aggregation
Answer: c) Lack of ADAMTS13 to cleave von Willebrand factor multimers
Explanation: Decreased ADAMTS13 activity → uncleaved vWF multimers → abnormal platelet adhesion → microthrombi → TTP (commonly in adult females).
🩸 Study Check 2 – Bernard–Soulier Syndrome
A 10-year-old boy presents with recurrent nosebleeds and easy bruising.
Exam reveals multiple petechiae on his legs.
Labs show normal PT/PTT, prolonged bleeding time, and low-normal platelet count.
Peripheral smear shows large platelets.
Which of the following best describes the underlying defect?
a) Autoantibodies against platelets causing splenic destruction
b) Deficiency of the GpIIb/IIIa receptor complex
✅ c) Deficiency of the GpIb receptor leading to defective platelet adhesion
d) Deficiency of ADAMTS13 enzyme
Answer: c) Deficiency of the GpIb receptor leading to defective platelet adhesion
Explanation: Bernard–Soulier syndrome is an autosomal recessive defect in the GpIb receptor, preventing platelets from binding vWF, impairing adhesion.
A 14-year-old girl presents with frequent nosebleeds and prolonged bleeding after minor cuts.
Labs show normal platelet count, normal PT/PTT, but prolonged bleeding time.
Platelet aggregation studies reveal failure to aggregate in response to ADP and fibrinogen.
Which of the following best explains these findings?
a) Autoantibodies against GpIb receptor
b) Decreased ADAMTS13 activity
c) Deficiency of GpIIb/IIIa receptors causing impaired platelet aggregation
d) Endothelial injury due to bacterial toxin
Answer: c) Deficiency of GpIIb/IIIa receptors causing impaired platelet aggregation
Explanation: Glanzmann Thrombasthenia is an autosomal recessive defect in the GpIIb/IIIa receptor, preventing platelets from binding fibrinogen → defective aggregation.