terma 3
1/100
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
101 Terms
egzergia (definicja)
praca maksymalna, gdy stanem końcowym przemiany jest stan równowagi układu i otoczenia o ściśle zdefiniowanych parametrach
Miara energii dostępnej zgromadzonej w układzie (potencjał układu do wykonania pracy)
egzergia (wzor)

sprawnosc egzergetyczna
procentowa miara nieodwracalności
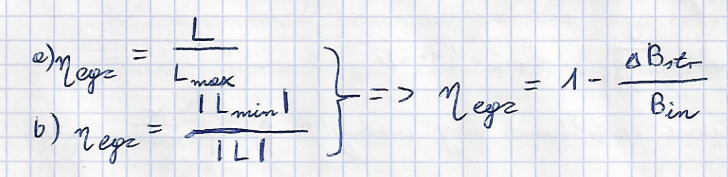
Praca (moc) maks./min.
praca (moc) uzyskana / doprowadzona w przemianie odwracalnej przy przejściu układu od stanu początkowego P do końcowego K
prawo gouy-stodoli
mowi o stracie pracy/ stracie egzergii
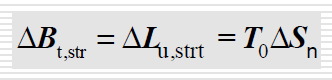
procesy nieodwracalne
dyssypacja energii, wymiana ciepla przy skonczonej roznicy temperatur, mieszanie, dlawienie pary wodnej
procesy odwracalne
kolejne stany układu i otoczenia w kierunku odwrotnym (od stanu końcowego do początkowego) są takie same, jak w procesie przechodzącym od stanu 1 do stanu 2.
zasada wzrostu entropii
Entropia układu (izolowanego) adiabatycznego nigdy nie maleje. Rośnie w przemianie nieodwracalnej, nie zmienia się w odwracalnej.
ZWE determinuje kierunek nieodwracalnych procesów w naturze takich jak: wymiana ciepła; przepływ wody z poziomu wyższego na niższy; dyfuzja masy wywołana różnicą koncentracji; reakcje chemiczne i przemiany fazowe; etc
sprawnosc wewnetrzna sprezarki
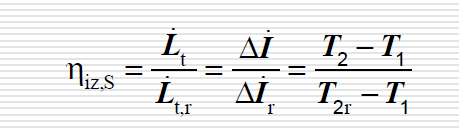
sprawnosc wewnetrzna turbiny
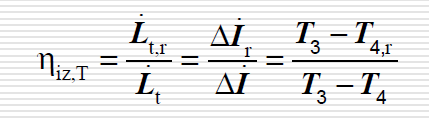
gaz wilgotny
mieszanina gazu suchego (niekondensującego) i H2O występującej w fazie gazowej (para), ciekłej lub stałej (lód).
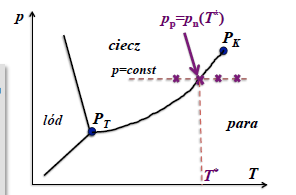
wykres fazowy H2O
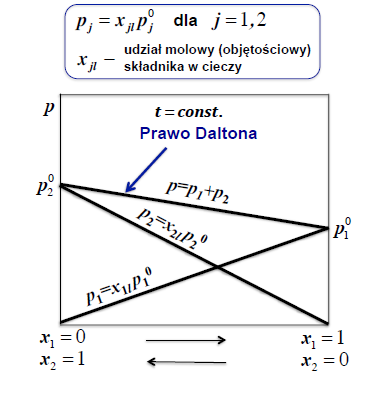
cisnienie czastkowe- PRAWO DALTONA
„Ciśnienie wywierane przez mieszaninę gazów jest równe sumie ciśnień wywieranych przez składniki mieszaniny, gdyby każdy z nich był umieszczany osobno w tych samych warunkach objętości i temperatury, jest ono zatem sumą ciśnień cząstkowych”.
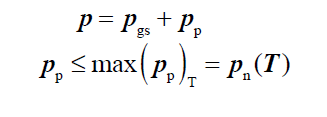
ZAWARTOŚĆ (STOPIEŃ) WILGOCI
stosunek masy pary do masy gazu suchego
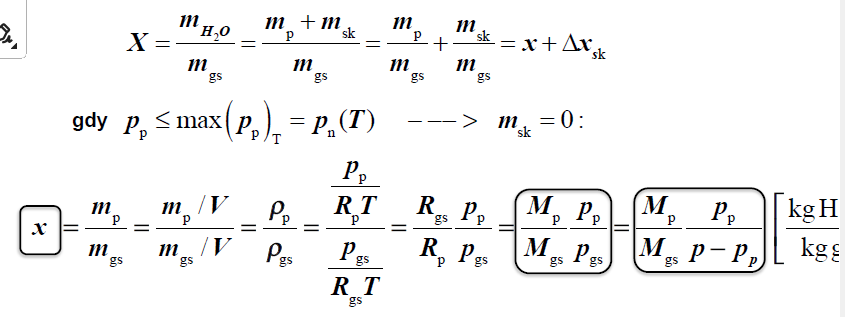
stopien nasycenia

wilgotnosc bezwzgledna
Gęstość pary wodnej przy jej ciśnieniu cząstkowym, p_p, w temperaturze mieszaniny, T
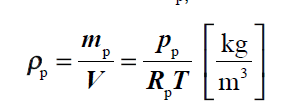
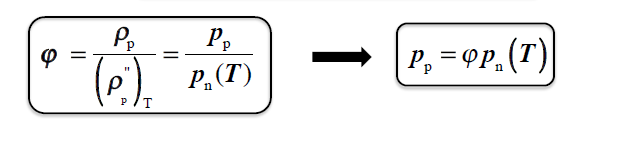
WILGOTNOŚĆ WZGLĘDNA
OBJĘTOŚĆ WŁAŚCIWA
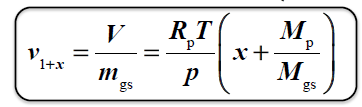
ENTALPIA WŁAŚCIWA
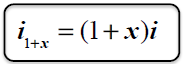
cisnienie czastkowe pary wodnej
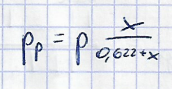
mozliwe stany gazu wilgotnego
-gaz wilgotny nienasycony (gaz suchy+przegrzana para wodna)
-gaz wilgotny nasycony (gaz suchy+para sucha nasycona
-gaz wilgotny dwufazowy, przesycony (gaz suchy+para sucha nasycona+skondensowana H2O)
gaz wilgotny nienasycony
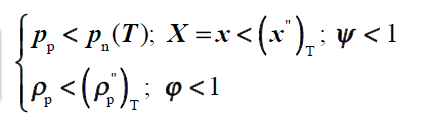
gaz wilgotny nasycony
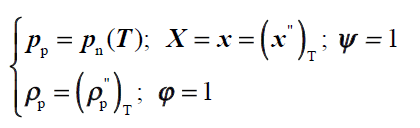
gaz wilgotny dwufazowy
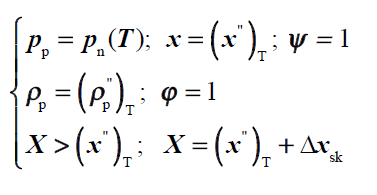
przemiany powietrza wilgotnego
-ogrzewanie, ochladzanie; x=const
-mieszanie izobaryczno-adiabatyczne
-nawilzanie izobaryczno-adiabatyczne
-osuszanie powietrza
ogrzewanie, ochladzanie
x=const
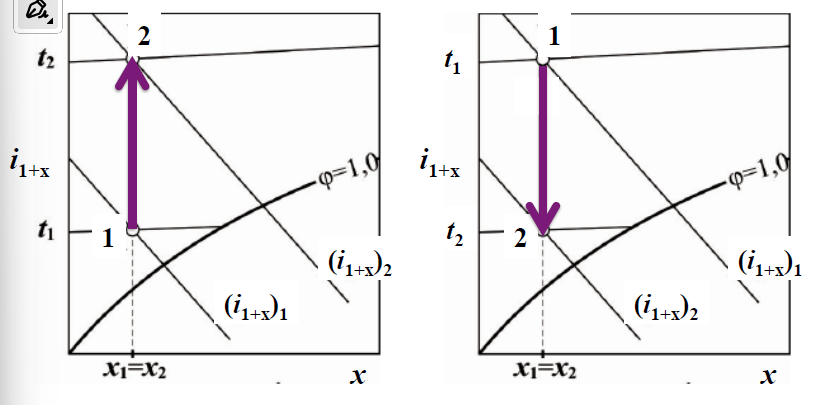
mieszanie izobaryczno-adiabatyczne
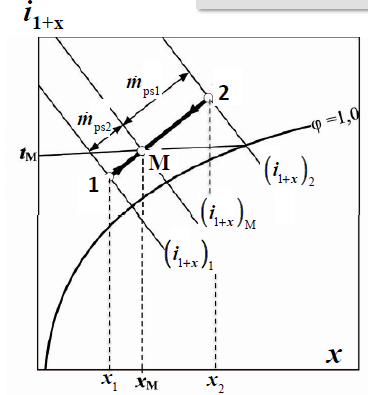
nawilzanie izobaryczno-adiabatyczne
osuszanie powietrza
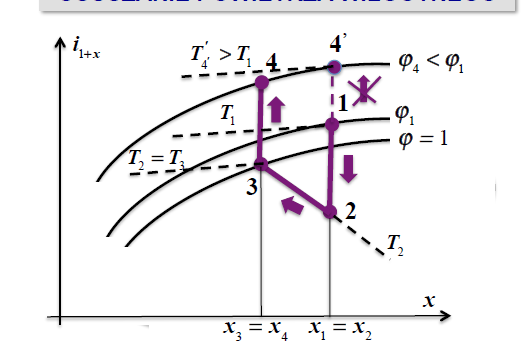
1-2 ochladzanie izobaryczne x=const
2-3 izotermiczne usuniecie skroplin
3-4 ogrzewanie izobaryczne x=const
Faza (definicja Gibbsa):
jednorodna pod względem właściwości część układu, oddzielona od pozostałej powierzchnią graniczną, na której niektóre właściwości substancji zmieniają się skokowo.
Substancja jednoskładnikowa może mieć tylko jedną fazę lotną, jedną fazę ciekłą i kilka faz stałych w postaci odmian alotropowych.
Przemiana fazowa (przejście fazowe)
przemiana termodynamiczna, w której następuje przejście jednej fazy w drugą, znajdującą się z nią w równowadze.
Zmiana parametru układu (np. p czy T) może prowadzić do pojawienie się nowej fazy.
rownowaga fazowa i jej warunki
-obie fazy mają jednakowe temperatury i ciśnienia (proces izobaryczno-izotermiczny).
-w stanie rownowagi termodynamicznej entalpia swobodna Gibbsa osiaga minimum dZ=0
-Równość swobodnych entalpii właściwych w warunkach równowagi termodynamicznej. z_alfa=z_beta
-entropia osiąga wartość maksymalną, tj. S = Smax, stąd: dS + dSo = 0
entalpia swobodna Gibbsa
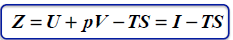
reguła faz Gibbsa
s=2+k-f
k-l. skladnikow
f-l. faz
s- l. stopni swobody
kierunek przebiegu przemiany fazowej
Faza o większej entalpii swobodnej właściwej przechodzi w fazę o mniejszej entalpii swobodnej właściwej.
wystepuja skokowe zmiany entropii i objetosci własciwej
efekt cieplny przemiany fazowej izotermicznej-izobarycznej
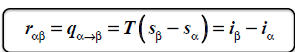
Wpływ ciśnienia na temperaturę przemiany fazowej – równanie Clapeyrona
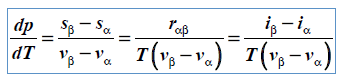
Równanie Clapeyrona dla pary wodnej
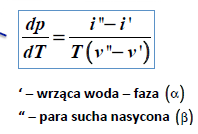
Równanie Clausiusa – Clapeyrona
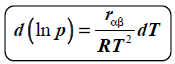
RÓWNOWAGA TERMODYNAMICZNA
stan w którym mierzalne własności układu wieloskładnikowego i wielofazowego nie zmieniają się w czasie. Obejmuje: równowagę mechaniczną, równowagę cieplną (termiczną), równowagę chemiczną oraz równowagę fazową.
MIESZANINA
układ złożony z dwóch lub większej liczby grupy cząstek o jednakowych własnościach fizycznych i chemicznych w całej objętości;
ROZTWÓR ROZCIEŃCZONY (ROZTWÓR)
mieszanina, w której jeden składnik (rozpuszczalnik) występuje w dużym nadmiarze w stosunku do drugiego (substancji rozpuszczonej), np. stopy metali, roztwory cieczy i gazu, dwóch ciał stałych.
parametry

mieszanina gazow doskonalych

funkcja calkowita (ukladu wieloskl.)
odniesiona do calego ukladu
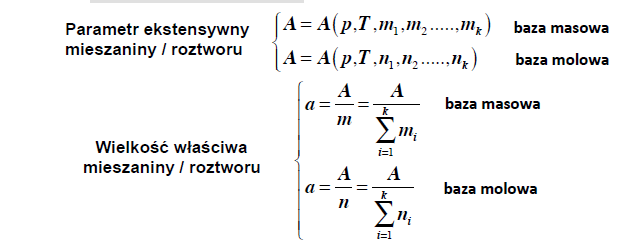
funkcja cząstkowa (ukladu wieloskl.)
odniesiona do ilosci i-tego skladnika

rownanie gibbsa-duhema

Związek pomiędzy molowymi wielkościami cząstkowymi i wielkością właściwą mieszaniny dla układu dwuskładnikowego

Proces mieszania dwoch skladnikow (nie roztwor doskonaly)
wystepuje efekt cieplny mieszania (rozpuszczania) i efekt zmiany objętości
Funkcja termodynamiczna roztworu jest sumą jej wartości dla czystych składników oraz efektu mieszania

Proces mieszania dwoch skladnikow (roztwor doskonaly)
Brak efektów ciepła mieszania i zmiany objętości
Każda wielkość cząstkowa równa jest odpowiedniej wielkości właściwej czystego składnika
Wielkości właściwe roztworu są równe ich sumie przed zmieszaniem
rozpuszczalnosc
nieograniczona: → roztwor doskonaly; wydzielanie ciepla mieszania
ograniczona: → pochlanianie ciepla mieszania
Ciśnienie cząstkowe składnika nad roztworem
jest mniejsze niż ciśnienie składnika nad jego czystą cieczą: p j < pj0
PRAWO RAOULTA
Liniowość zmian ciśnień cząstkowych par składników roztworu w funkcji udziałów molowych tych składników w roztworze ciekłym.
Skład pary nad roztworem jest inny niż skład roztworu ciekłego. Dla roztworu doskonałego wyznaczamy go z praw Raoulta oraz Daltona
W stałej temperaturze ciśnienie pary nasyconej składnika (prężność składnika) jest wprost proporcjonalne do ułamka molowego składnika w roztworze
ochylenia od prawa raoulta - roztwor rzeczywisty
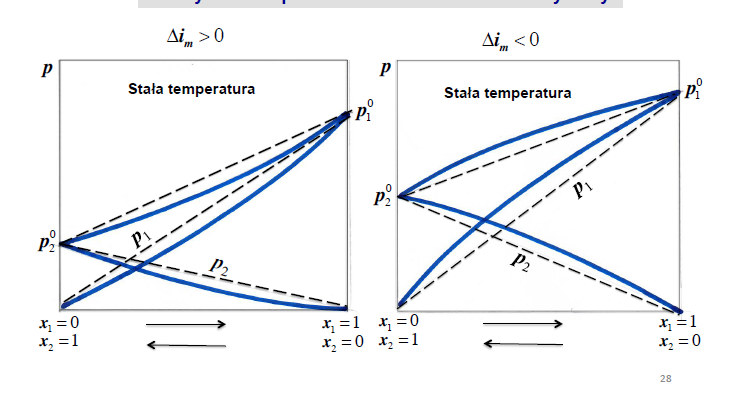
ochylenia od prawa raoulta - roztwor rozcienczony
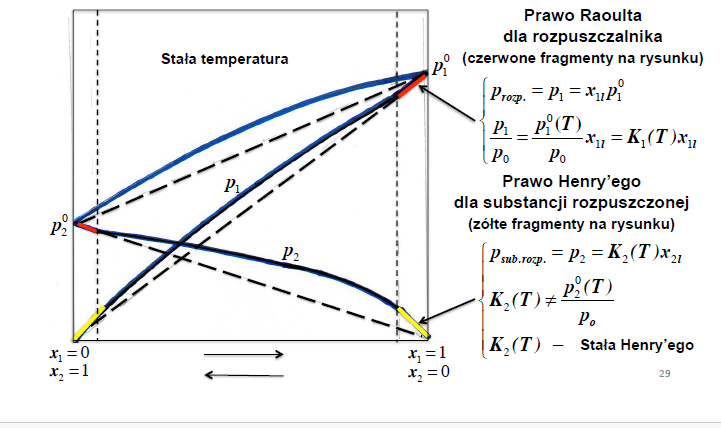
Prawo Henry’ego dla substancji rozpuszczonej
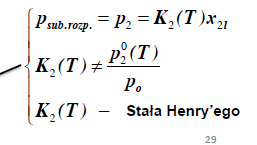
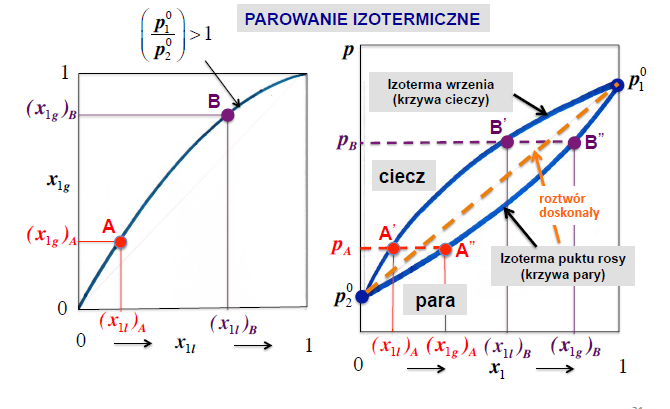
parowanie izotermiczne
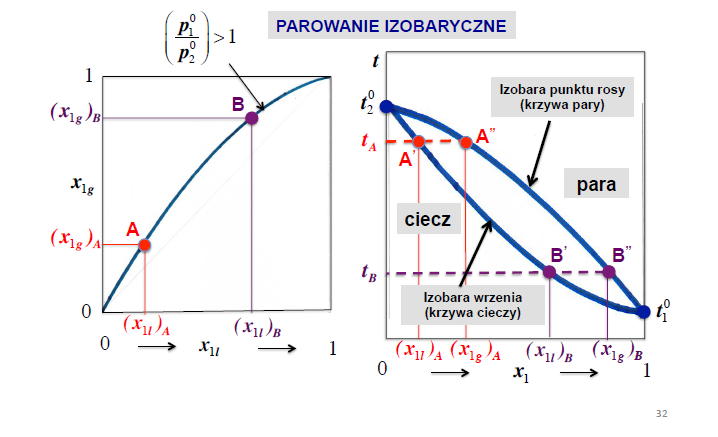
parowanie izobaryczne
Destylacja prosta
odparowanie cieczy, a następnie oziębienie jej par i skroplenie. Wykorzystanie różnicy między składem pary i cieczy (prawo Raoulta). Uzyskiwanie roztworu bogatszego w składnik bardziej lotny.
Destylacja frakcyjna (rektyfikacja)
wielokrotna destylacja prosta prowadzona w kolumnach rektyfikacyjnych. Zastosowania: w przemyśle naftowym, spirytusowym, farmaceutycznym.
zeotrop
roztwór wykazujący niewielkie odchylenia od prawa Raoulta; można go rozdzielić na składniki na drodze destylacji; skład roztworu w każdej temperaturze (przy każdym ciśnieniu) jest inny niż skład pary nad roztworem.
AZEOTROP
roztwór wykazujący znaczne odchylenia od prawa Raoulta; nie może być rozdzielony na składniki na drodze destylacji
PUNKT AZEOTROPOWY – taki sam skład pary i cieczy
AZEOTROP DODATNI
Maksymalne ciśnienie całkowite roztworu większe od prężności czystego bardziej lotnego składnika. Minimalna temperatura roztworu mniejsza od temperatury wrzenia czystego bardziej lotnego składnika.
AZEOTROP UJEMNY
Minimalne ciśnienie całkowite roztworu mniejsze od prężności czystego mniej lotnego składnika. Maksymalna temperatura roztworu większa od temperatury wrzenia czystego mniej lotnego składnika.
Rozpuszczalność
maksymalna ilość substancji (w gramach lub molach) którą można rozpuścić w danej ilości rozpuszczalnika (w kg lub m3) by uzyskać roztwór nasycony.
Rozpuszczalność fazy stałej w cieczy
• Faza stała rozpuszcza się w cieczy w sposób ograniczony, tworząc roztwór rozcieńczony.
• Rozpuszczalność zależy od temperatury przy danym ciśnieniu.
• Maksymalna ilość substancji stałej jaka może się rozpuścić w cieczy, tworząc jednofazowy roztwór ciekły, określana jest mianem rozpuszczalności.
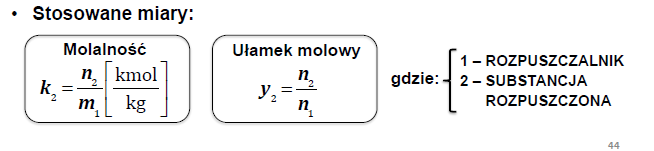
2 prawo raoulta
Podwyższenie temperatury wrzenia roztworu/obnizenie temperatury krzepniecia
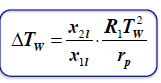
STAŁA EBULIOSKOPOWA
przyrost temperatury wrzenia roztworu zawierającego 1 mol substancji rozpuszczonej w 1 kg rozpuszczalnika.
r_p - cieplo parowania
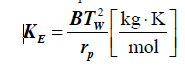
STAŁA KRIOSKOPOWA
spadek temperatury krzepnięcia roztworu zawierającego 1 mol substancji rozpuszczonej w 1 kg rozpuszczalnika.
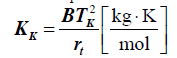
Zasada zachowania masy w reakcji chem.
W reakcji chemicznej zachowana jest równość mas substratów i produktów, nie oznacza zachowania liczby moli reagentów - w wyniku reakcji może ich ubywać (kompresja) lub przybywać (ekspansja).
ENTALPIA REAKCJI W STANIE STANDARDOWYM
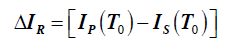
Entalpia tworzenia
Entalpia tworzenia – efekt cieplny reakcji syntezy jednego kilomola związku w stanie standardowym (wyznaczany eksperymentalnie, tablice)
Entalpia tworzenia prostych pierwiastków (np. O2, N2, H2) będących w trwałej postaci w stanie standardowym jest równa zeru.
Efekt cieplny reakcji chemicznej
Największa możliwa ilość ciepła otrzymana lub pochłonięta w reakcji izobaryczno-izotermicznej (przepływowa komora spalania) lub izochoryczno-izotermicznej (zamknięta komora spalania)
Termodynamiczna konwencja znakowa
+ cieplo doprowadzone → reakcja endotermiczna
- cieplo odprowadzone → reakcja egzotermiczna
PRAWO HESSA
niezależność efektu cieplnego reakcji od drogi przemiany
Ciepło reakcji chemicznej, przebiegającej w stałej objętości lub pod stałym ciśnieniem nie zależy od drogi reakcji chemicznej, a jedynie od stanu początkowego i końcowego; nie zależy od tego, czy produkty otrzymano z substratów w bezpośredniej reakcji czy poprzez dowolne reakcje pośrednie.
PRAWO HESSA (PRAWO STAŁEJ SUMY CIEPEŁ):
Suma efektów cieplnych następstwa reakcji izotermiczno-izobarycznej lub izotermiczno-izochorycznej jest równa sumie efektów cieplnych dowolnych kolejnych reakcji o tych samych substancjach wyjściowych i tych samych substancjach końcowych oraz przy tych samych p,T lub v,T.
PRAWO KIRCHOFFA
zależność efektu cieplnego reakcji od temperatury
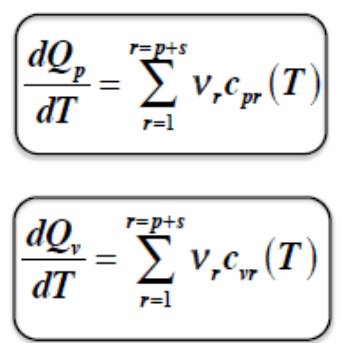
RÓWNOWAGA CHEMICZNA
stan, w którym stosunki ilościowe substratów i produktów nie ulegają zmianie.
RÓWNOWAGA TERMODYNAMICZNA (z czego sie sklada)
RÓWNOWAGA CHEMICZNA + RÓWNOWAGA MECHANICZNA (stałe p) +RÓWNOWAGA CIEPLNA (stałe T )
stała szybkości reakcji prostej (syntezy)
szybkość spadku koncentracji substratów w reakcji od lewej do prawej
stała szybkości reakcji odwrotnej (dysocjacji)
szybkość wzrostu koncentracji substratów w reakcji od prawej do lewej
LICZBA POSTĘPU REAKCJI
ζ = 0 → brak reakcji, jeszcze się nie rozpoczęła
ζ = 1 → przereagowały stechiometryczne liczby moli, stan rownowagi
Kierunek przebiegu reakcji wynika z II ZT:
reakcja zachodzi tak by: ΔZR = ZP − ZS < 0 → ZP < ZS
POTENCJAŁ CHEMICZNY
zmiana energii wewnętrznej U ze zmianą ilości moli reagenta ni, przy stałych V i S oraz niezmiennych liczbach moli pozostałych związków.
Potencjał chemiczny równy jest cząstkowej entalpii swobodnej składnika.
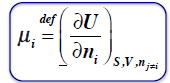
RÓWNANIE GIBBSA
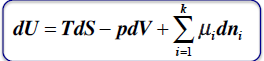
Cząstkowa entalpia swobodna
wyraża zmianę swobodnej entalpii składnika układu względem ilości tego składnika, gdy T i p są stałe oraz gdy ilości innych składników nie zmieniają się.
POWINOWACTWO CHEMICZNE REAKCJI
gdy stan rownowagi to A=0
reakcja zachodzi samorzutnie gdy A>0
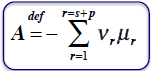
Kierunek reakcji chemicznej na podstawie znaku Lpmax
Równanie Helmholtza Gibbsa
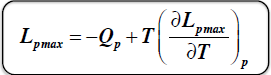
Ciśnieniowa stała równowagi chemicznej
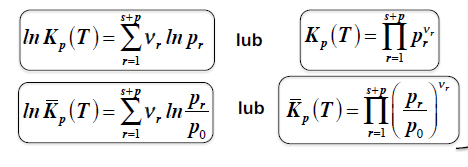
IZOTERMA VAN’T HOFFA
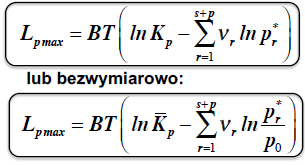
DYSOCJACJA
rozkład cząstek związku lub pierwiastków chemicznych na składniki prostsze w wysokich temperaturach. Reakcja endotermiczna.
STOPIEŃ DYSOCJACJI α
stosunek liczby moli związku chemicznego, która uległa rozkładowi w stanie równowagi do początkowej liczby moli n0 tej substancji dysocjującej
Jonizacja
utrata elektronów przez atomy: A⇔ A+ + e−
w bardzo wysokich temperaturach (łuk elektryczny, kilka tysięcy K) lub w umiarkowanych temperaturach przy niskich ciśnieniach, np. w górnej warstwie atmosfery.
Zakładając równowagę cieplną w plazmie, jonizację można traktować jako reakcję chemiczną, w której powstaje mieszanina doskonała atomów, dodatnich jonów i elektronów.
TEOREMAT NERNSTA – III ZT
W reakcjach chemicznych zachodzących w układach skondensowanych przy p=const. i T=const., krzywe zależności efektu cieplnego reakcji oraz pracy maksymalnej od temperatury zbiegają się do wspólnej stycznej w pobliżu temperatury zera bezwzględnego.
Entropie molowe reagentów dążą do zera, gdy temperatura zbliża się do 0K.
Dla ciała skondensowanego molowe ciepła reagentów dążą do zera w pobliżu temperatury zera bezwzględnego
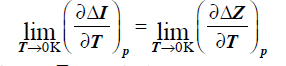
POSTULAT PLANCKA
Entropia jednorodnego chemicznie ciała o skończonej gęstości znajdującego się w stanie równowagi trwałej, przy zmniejszaniu temperatury bezwzględnej do zera, zbliża się do zerowej wartości granicznej
KINETYKA CHEMICZNA
analiza zmiany stężeń substratów i produktów reakcji chemicznej w czasie, badanie szybkości reakcji w dążeniu do stanu równowagi.
SZYBKOŚĆ REAKCJI
zmiana stężenia reagenta w czasie:

RÓWNANIE KINETYCZNE
RÓWNANIE KINETYCZNE – opisuje zmiany stężenia molowego
substratu, s, w czasie reakcji:
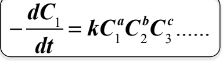
RZĄD REAKCJI
liczba składników, których stężenie decyduje o szybkości reakcji = sumie wykładników potęgowych równania kinetycznego.