Neurotransmitters System I: Glutamate
1/15
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
16 Terms
What key requirements must a molecule meet to be considered a neurotransmitter?
It must be synthesised in the presynaptic neuron.
It must be stored in presynaptic vesicles.
It must be released from the axon terminal upon stimulation (usually Ca²⁺-dependent).
It must bind and activate postsynaptic receptors, producing a response.
A neurotransmitter also requires a mechanism for removal (reuptake or enzymatic degradation) and must produce reproducible physiological effects when experimentally applied.
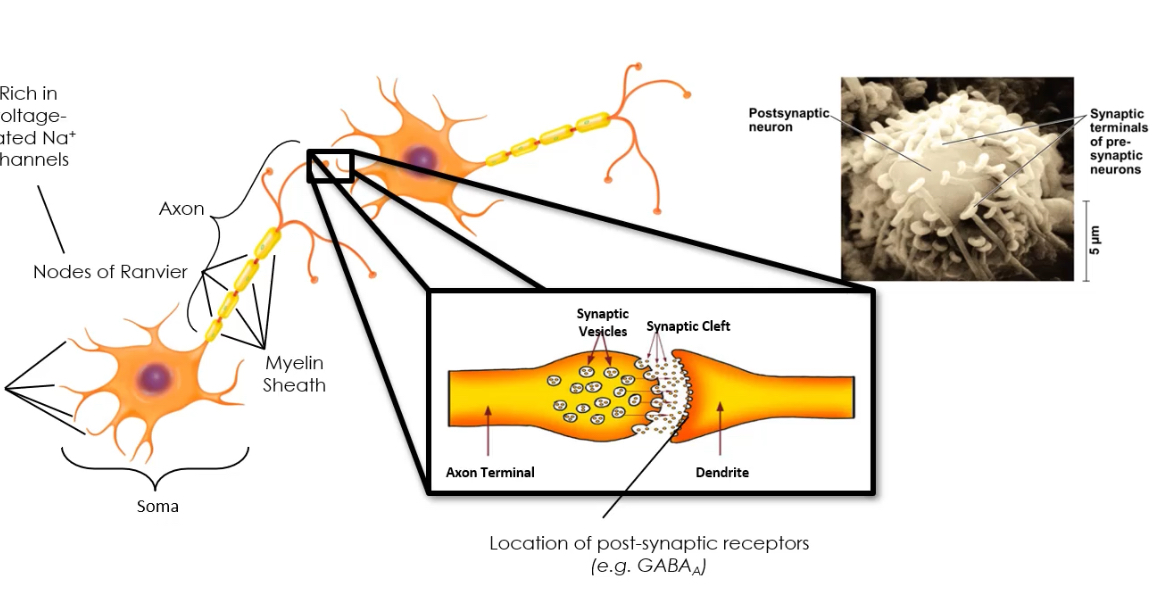
What are the steps of neurotransmission?
1. Neurotransmitter synthesis (soma or terminal).
2. Loading into vesicles.
3. Vesicle fusion and release after AP-evoked Ca²⁺ entry.
4. Binding to receptors on the postsynaptic cell.
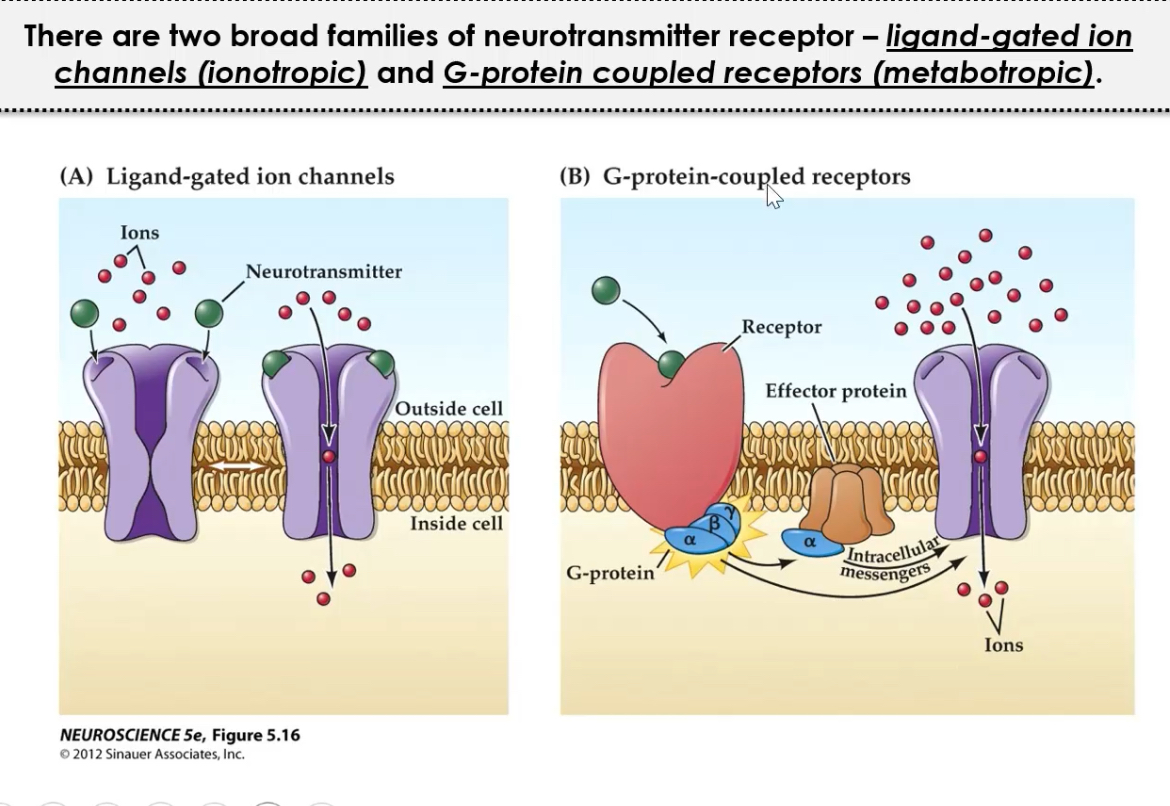
Receptors may be ionotropic or metabotropic, enabling fast or slow signalling respectively.
What is glutamate’s major role in the CNS?
It is the major excitatory neurotransmitter in the CNS responsible for the majority of fast excitatory synaptic transmission.
Why was glutamate slow to be recognised as a neurotransmitter?
It sits at metabolic crossroads, participating in multiple pathways (TCA cycle, amino acid metabolism), making it hard to prove its signalling-specific role.
Glutamate exists at high concentrations in all cells, so demonstrating vesicular release and synaptic specificity required advanced methods.
How is glutamate synthesised and stored in neurons?
1. From glutamine (major pathway):
Glutamine → Glutamate
Enzyme: Glutaminase
Occurs in nerve terminals.
2. From α-ketoglutarate:
Via transamination reactions in the TCA cycle.
Glutamine is supplied by astrocytes → core of the glutamate–glutamine cycle, preventing excitotoxicity.
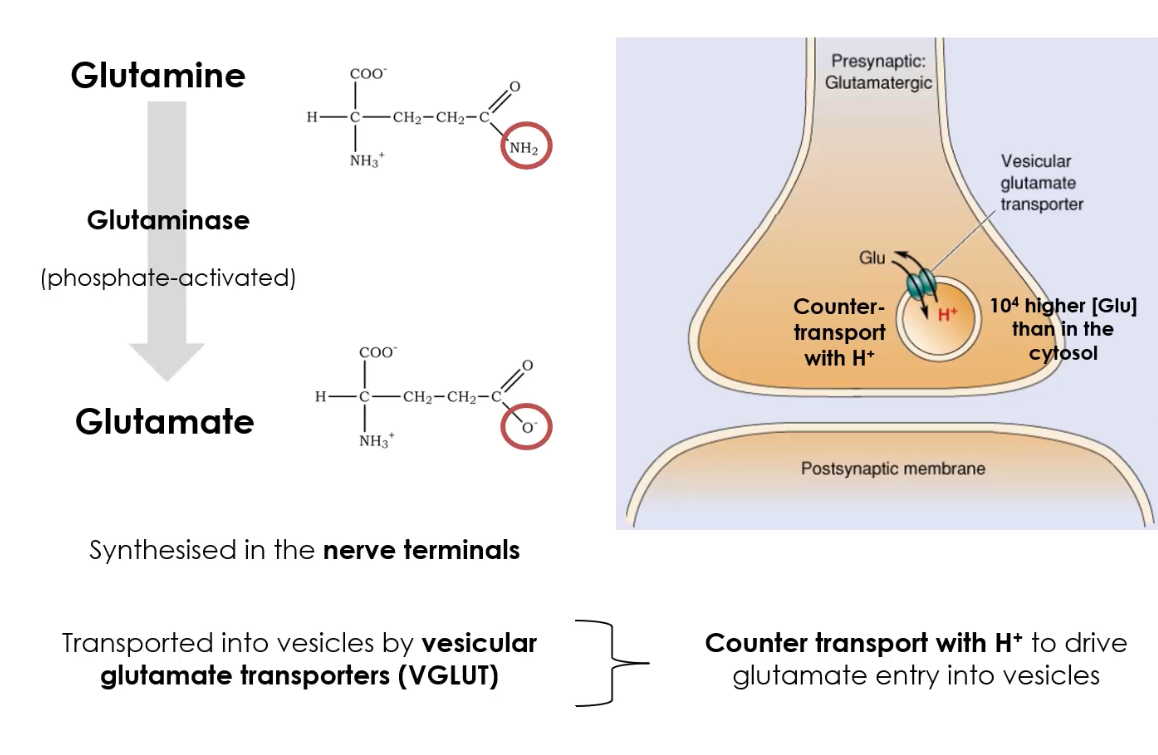
How is glutamate stored before release?
Loaded into synaptic vesicles by Vesicular Glutamate Transporters (VGLUTs).
VGLUTs maintain high intravesicular concentrations so that vesicles release quantal packets of glutamate.
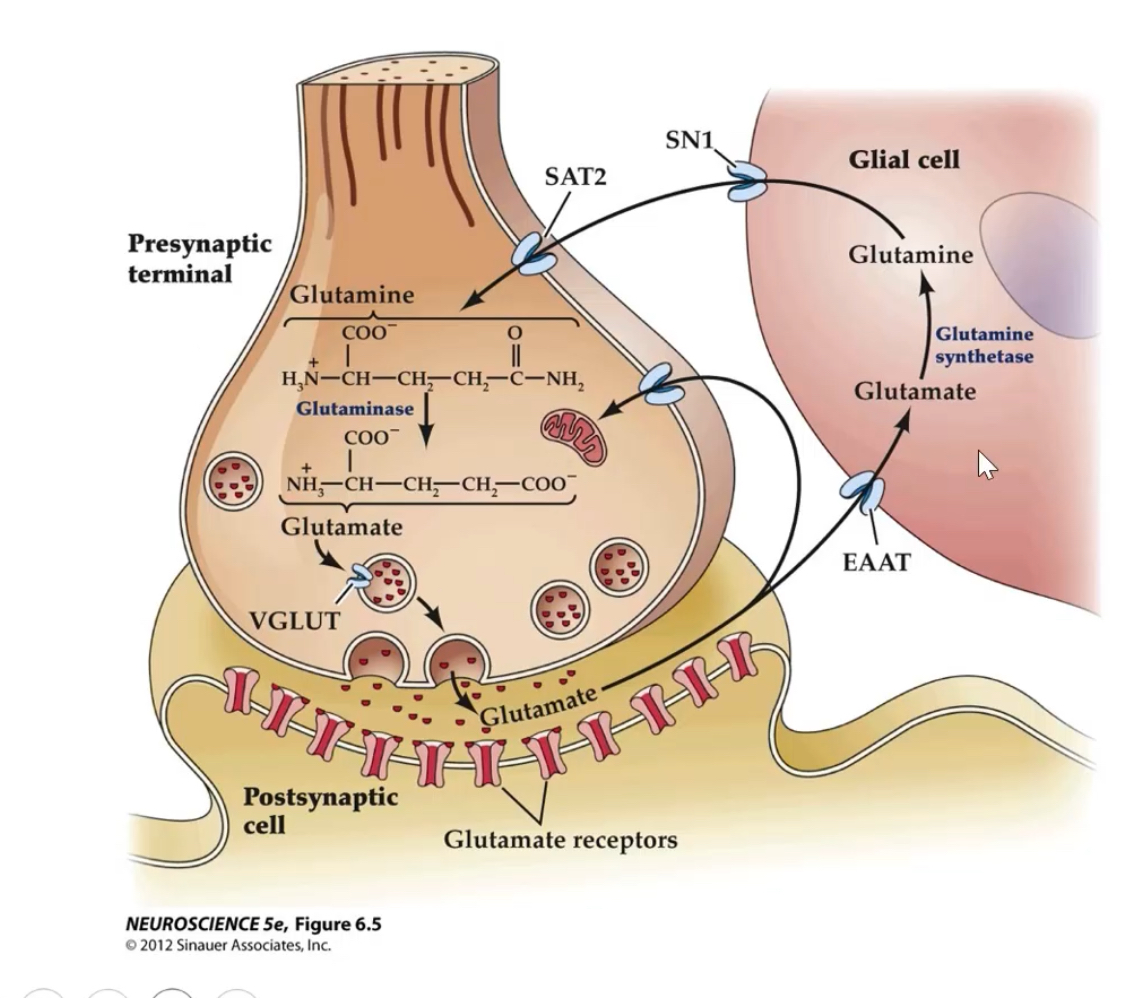
How is extracellular glutamate removed and recycled?
Glutamate is released
An action potential arrives at the presynaptic terminal → glutamate is released into the synaptic cleft.Glutamate must be cleared quickly
Glutamate is excitatory, so if it stays in the cleft it would keep activating receptors and cause excitotoxicity.Re-uptake via EAATs
Excitatory Amino Acid Transporters (EAATs) on:Astrocytes (mainly EAAT2 / GLT-1)
Presynaptic neurons
actively remove glutamate from the synaptic cleft.
Conversion in astrocytes (degradation step)
Inside astrocytes, glutamate is not reused directly.
It is converted to glutamine by the enzyme glutamine synthetase.
(Glutamate → Glutamine)Transport back to neurons
Glutamine is released from astrocytes via transporters (e.g. SN1 / SAT2) and taken up by neurons.Recycling in neurons
In neurons, glutamine is converted back into glutamate, which is repackaged into vesicles for future neurotransmission.
EAAT2 (GLT-1) is the main glial transporter; dysfunction contributes to ALS and epilepsy.
What are the two major families of glutamate receptors?
1. Ionotropic (ligand-gated ion channels):
AMPA, NMDA, Kainate
2. Metabotropic (G-protein coupled):
mGluR Groups I, II, III
Ionotropic receptors mediate fast EPSCs; metabotropic receptors modulate excitability and plasticity.
Ionotropic glutamate receptors
Named after the agonists that activate them
AMPA and NMDA receptors are majority of excitatory neurotransmission in the brain
AMPA receptors
AMPA receptors are ionotropic glutamate receptors that mediate fast excitatory synaptic transmission in the CNS.
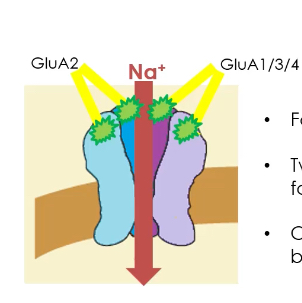
They are made from four subunits, chosen from GluA1, GluA2, GluA3, GluA4 (including splice variants).
The receptor is heterotetrameric, arranged as a “dimer of dimers.”
Each receptor has four glutamate binding (orthosteric) sites.
At least two sites must be occupied by glutamate for the channel to open.
As more binding sites are occupied, the ionic current increases.
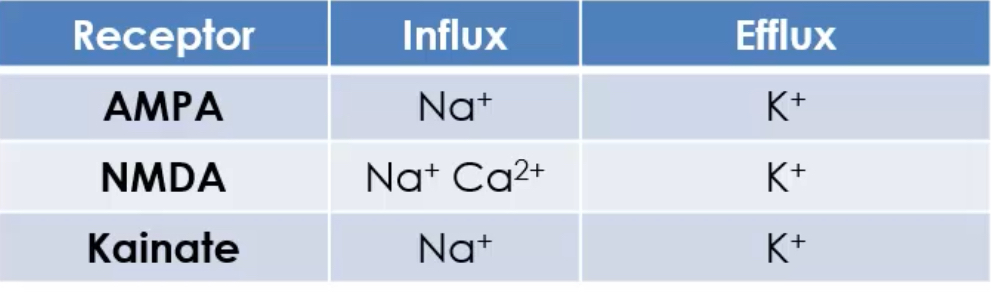
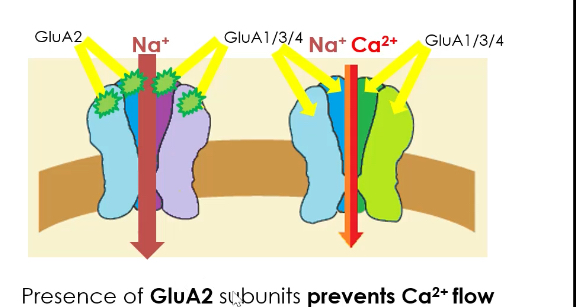
Ion permeability depends on GluA2:
If GluA2 is present, the channel is Na⁺ permeable only.
If GluA2 is absent, the channel allows Na⁺ and Ca²⁺ to enter.
Most common AMPA receptor composition:
2 × GluA2 subunits
2 × GluA1, GluA3, or GluA4 subunits
Functional consequence:
Presence of GluA2 prevents Ca²⁺ influx, protecting neurons from excitotoxicity.
Lack of GluA2 makes AMPA receptors Ca²⁺-permeable, which is important in plasticity but increases excitotoxic risk.
NMDA receptors
NMDA receptors are ionotropic glutamate receptors involved in synaptic plasticity, learning, and memory.
Slower onset, longer lasting.
Conduct significant Ca²⁺, triggering intracellular signalling.
They are composed of three possible subunit types: GluN1 (NR1), GluN2 (NR2), GluN3 (NR3), with splice variants.
The receptor is heterotetrameric, arranged as a dimer of dimers.
Most common composition:
2 × GluN1 subunits
2 × GluN2 subunits (sometimes GluN3 instead)
Dual ligand requirement:
Glutamate binds to GluN2
Glycine or D-serine binds to GluN1
All ligand-binding sites must be occupied for activation
Voltage dependence:
At resting membrane potential, the channel pore is blocked by Mg²⁺
Postsynaptic depolarisation expels Mg²⁺ from the pore
Channel opening requires two conditions simultaneously:
Ligand binding (glutamate + glycine/D-serine)
Membrane depolarisation to remove the Mg²⁺ block
Ion permeability:
NMDA receptors allow Ca²⁺, Na⁺, and K⁺ to pass
Ca²⁺ influx is the key signal for synaptic plasticity (e.g. LTP)
Role of GluN3:
Inclusion of GluN3 subunits reduces receptor activity
Functionally inhibitory/modulatory to NMDA receptor signalling

Kainate Receptors
Kainate receptors are ionotropic glutamate receptors that mediate excitatory neurotransmission, but with more modulatory roles than AMPA receptors.
• They are built from five possible subunits: GluK1–GluK5
• The functional receptor is tetrameric.
• Subunit assembly rules:
• GluK1–GluK3 can form homomeric receptors or heteromeric receptors with each other.
• GluK4 and GluK5 cannot form functional receptors alone.
• GluK4/5 must combine with GluK1–3 to form functional heteromeric channels.
Gating mechanism:
• They are ligand-gated ion channels.
• Glutamate binding is required for channel opening.
• The exact gating and signalling mechanisms are less well understood than for AMPA and NMDA receptors.
Distribution and function:
• Kainate receptors have a more limited distribution in the brain compared with AMPA and NMDA receptors.
• They often act at presynaptic terminals, where they modulate neurotransmitter release, rather than producing large fast EPSPs.
What are the three groups of mGluRs and their main features?
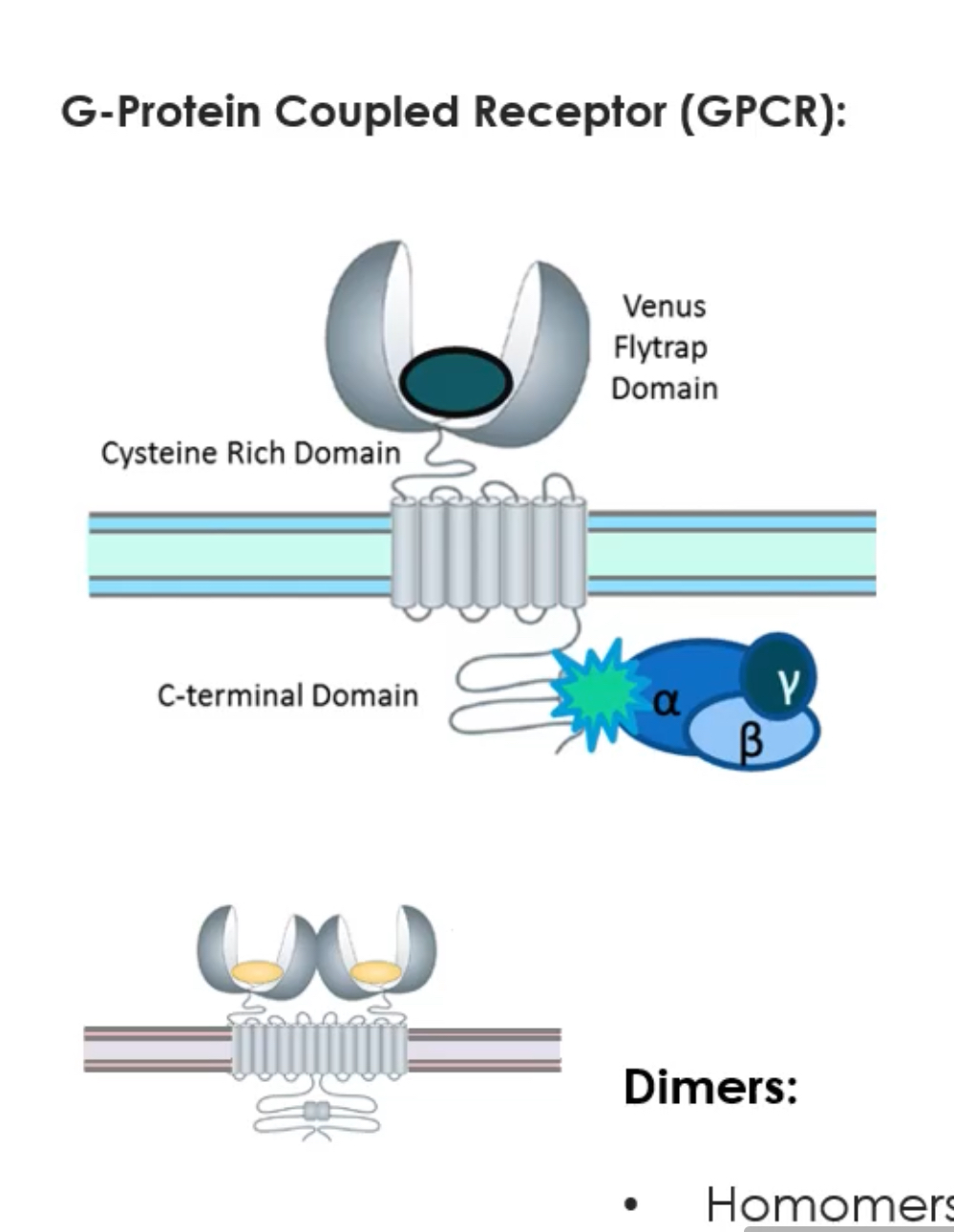
Metabotropic glutamate receptors (mGluRs) are G-protein coupled receptors (GPCRs), not ion channels.
When glutamate binds, they do not open a pore.
Instead, they activate G-proteins, which trigger intracellular signalling pathways.
Their effects are slower, longer-lasting, and modulatory compared to AMPA/NMDA receptors.
Structure
Each receptor has a “Venus flytrap” domain outside the cell where glutamate binds.
They span the membrane 7 times (typical GPCR).
They function as dimers (two receptors together).
Dimers can be homomers (same subtype) or heteromers (different receptors).
There are 8 subtypes, divided into 3 functional groups:
Group 1: mGlu1 and mGlu5
Usually postsynaptic
Coupled to Gq proteins
Activate PLC → IP₃ + DAG
IP₃ releases Ca²⁺ from the ER
Effect: increase postsynaptic excitability and support synaptic plasticity
Group 2: mGlu2 and mGlu3
Usually presynaptic
Coupled to Gi/o proteins
Inhibit adenylyl cyclase → ↓ cAMP
Effect: reduce neurotransmitter release
Group 3: mGlu4, mGlu6, mGlu7, mGlu8
Usually presynaptic
Also Gi/o coupled
↓ cAMP
Effect: strong inhibition of neurotransmitter release
Group 1 = postsynaptic → excitatory/modulatory
Groups 2 & 3 = presynaptic → inhibitory/modulatory
What is the difference between an EPSC and an EPSP?
EPSC: Excitory Post Synaptic Current
The flow of ions, and change in current across a post synaptic membrane
EPSCs lead to the generation of Excitatory Post Synaptic Potential(EPSPs) which increase likelihood of firing an action potential
EPSP: Excitory Post Synaptic Potential
Change in membrane voltage caused by the EPSC.
EPSCs via NMDA and Kainate receptors are slower and longer-lived than AMPA-mediated EPSCs.
What is excitotoxicity?
Pathological process where excessive excitatory stimulation (especially Ca²⁺ influx via NMDA) leads to neuronal injury and death.
Seen in stroke, traumatic brain injury, and Alzheimer’s disease where glutamate clearance is impaired.
Define long-term potentiation (LTP).
Persistent strengthening of synaptic transmission following repeated patterns of activity.
LTP is a cellular correlate of learning and memory.
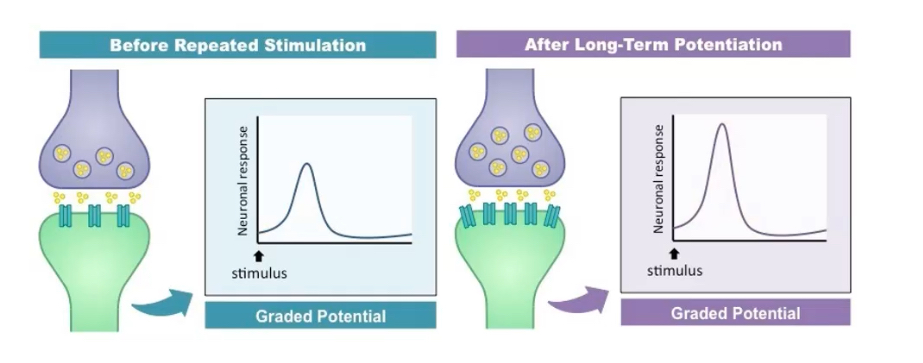
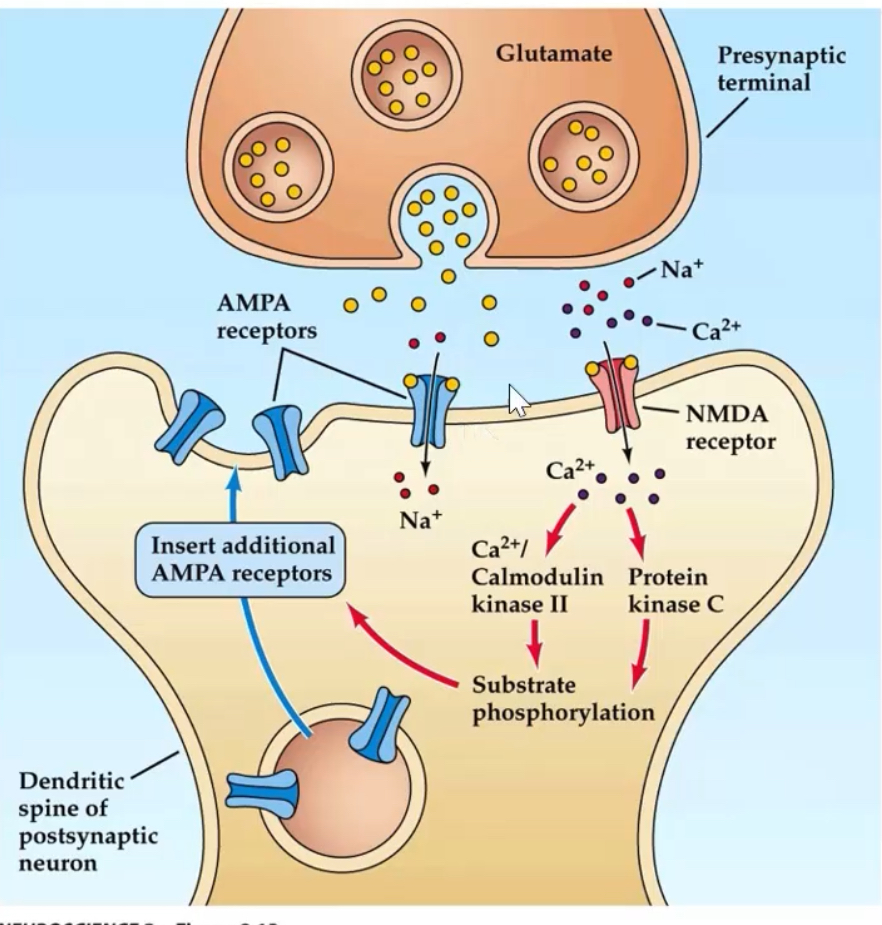
What are the molecular steps underlying early-phase LTP?
1. Glutamate activates AMPA receptors → Na⁺ influx → depolarisation.
2. Depolarisation removes Mg²⁺ block from NMDA receptors.
3. NMDA receptors open → Ca²⁺ influx.
4. Ca²⁺ activates Calmodulin kinase II and Protein Kinase C
5. These kinases promote insertion of new AMPA receptors into postsynaptic membrane.
6. Result: increased AMPA receptor number and conductance → stronger synaptic response.
CaMKII autophosphorylation provides a molecular “memory,” stabilising the potentiated state.
Without Ca²⁺, synapses cannot enter a potentiated state even if glutamate is present.