(cls 646) leukemias, lymphomas, & lipid storage disorders
1/69
Earn XP
Description and Tags
also includes lymphoproliferative disorders
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
70 Terms
overview of leukemia
progressive, malignant disease of the hematopoietic system characterized by unregulated, clonal proliferation of the hematopoietic stem cells
malignant cells usually replace normal marrow cells and eventually interfere with normal BM function
cells may invade other organs; if patient is not treated. leukemic cells eventually cause death
generally classified according to whether they are acute or chronic diseases, based on the aggressiveness of the illness
overview of acute leukemia
unregulated, immature, undifferentiated or minimally differentiated malignant cells with a gap in the normal maturation stages (i.e., "leukemic hiatus")
aggressive; abrupt onset of symptoms (fever, hemorrhage, weakness)
affects all ages
WBC blasts constitute at least 20% of all nucleated marrow cells
WBC counts may be elevated, normal, or low
hallmark signs: anemia, neutropenia, and thrombocytopenia
mild organomegaly (spleen, liver)
death within months if treatment is not begun
overview of chronic leukemia
less aggressive; insidious onset of symptoms (weakness, pallor, severely enlarged spleen & liver)
dx often made during routine physical following investigation of nonspecific complaints
adults usually affected
WBC usually elevated; differential is similar in both the peripheral blood and marrow
BM infiltrated by increased number of mature cells
anemia often present, but platelets are normal or increased; thrombocytopenia rare until late in the disease
all stages of maturation in granulocytic leukemia
progresses slowly and death occurs years after diagnosis
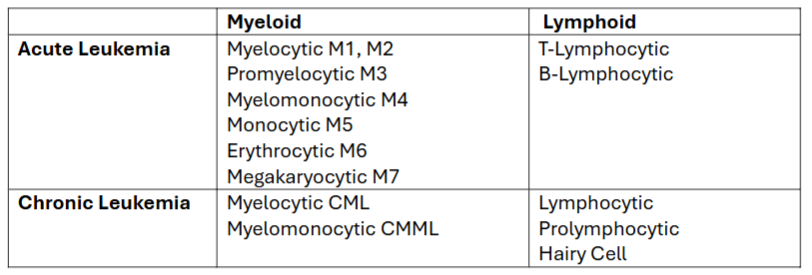
general classification of acute/chronic leukemias
can each be further subgrouped according to the stem cell from which the abnormal clone of cells differentiated (see table)
if cells were derived from the CFU-GEMM → myeloid leukemia
if cells were derived from the CFU-lymphoid → lymphocytic leukemia
etiology/pathophysiology of leukemia
cause is not clear; factors thought to play a role in development of leukemia:
radiation; drugs/chemicals (chloramphenicol, phenylbutazone, arsenic-containing compounds, sulfonamides, some insecticides)
genetics; down’s syndrome; fanconis syndrome
bloom syndrome (chr breakage/rearrangement)
viruses
***abnormal clone arises from a somatic mutation of one HSC; as that cell proliferates it crowds out the normal cells in BM and begins to spill over into peripheral blood
acute myeloid leukemia (AML) presentation
pallor, lethargy, fatigue, dyspnea, and weakness from anemia
bleeding, bruising, and petechial hemorrhages, purpura, epistaxis, and gingival bleeding caused by thrombocytopenia
infections fail to respond to treatment, fever related to infection caused by neutropenia
organ infiltration: bone tenderness, splenomegaly, hepatomegaly lymphadenopathy, gum hypertrophy, skin infiltrates, meningeal syndrome (headache, nausea, vomiting)
AML lab findings (rbcs, plts, coag, wbc)
RBCs
Normochromic, normocytic anemia
nRBC, aniso, poiks
Platelets: decreased; hypogranular, giant
Coagulation: defects may be present
WBCs: Usually increased
Peripheral blood blasts: 15–95%
Auer rods may be present
Monocytosis; wariable eosino/basophilia
Neutropenia with dysplastic neutrophils
AML lab findings (BM, cytogenetics)
Bone Marrow:
Hypercellular with dysplastic cells
Blasts >20%
Cytogenetic abnormalities:
Approx 70% have clonal acquired chromosomal abnormalities that can be single, numerical or structural
Trisomy 8 common in AML but not diagnostic for a specific type
(characteristics of AML FAB classification) AML-M0
undifferentiated AML
blasts: >30%
cytogenetic abnormalities: variable structural and numeric aberrations
cell markers: HLA-DR+, MPO+, CD13+, 33+
(characteristics of AML FAB classification) AML-M1
AML with minimal maturation
blasts: >90%; myeloid <10%, monocytic <10%
cytogenetic abnormalities: same as M0
cell markers: HLA-DR+, MPO+, CD13+, 33+
(characteristics of AML FAB classification) AML-M2
AML with maturation
blasts: 30-89%; myeloid >10%, monocytic <20%
cytogenetic abnormalities: t(8:21)(q22;q22) in 15% of cases
cell markers: CD19+
(characteristics of AML FAB classification) AML-M3
acute promyelocytic leukemia (APL)
blasts: <30%; promyelos >50%
cytogenetic abnormalities: t(15;17)(q22:q11.2) in >90% of cases
cell markers: HLA-DR-, CD2+
(characteristics of AML FAB classification) AML-M4
acute myelomonocytic leukemia (AMML)
blasts: >30%; myeloid/monocytic >20%;
AML-M4 eo = >5% eos
cytogenetic abnormalities: abnormal 11q, variable aberrations
inv(16)(p32;q22)
t(16;16), del(16)(q22)
cell markers: CD11c+, 14+, 2+
(characteristics of AML FAB classification) AML-M5
acute monocytic leukemia
blasts
M5a >80% ; M5b <80%
cytogenetic abnormalities:
t(9;11)(p22;q23)
abnormal 11q t(8:16)
cell markers: CD11c+, 14+
(characteristics of AML FAB classification) AML-M6
acute erythroid leukemia
blasts >30%; erythroid >50%
cytogenetic abnormalities: n/a
cell markers: glycophorin A+
(characteristics of AML FAB classification) AML-M7
acute megakaryocytic leukemia
blasts: >30%
cytogenetic abnormalities: none
cell markers: CD41+, 61+

(staining patterns of AML) undifferentiated AML (M0)
MPO/SB ; SE ; NSE -
PAS +
(staining patterns of AML) AML w minimal maturation (M1)
MPO/SB +
SE ; NSE ; PAS -
(staining patterns of AML) AML with maturation (M2)
MPO/SB ; SE +
NSE -
PAS -/diffuse
(staining patterns of AML) acute promyelocytic leukemia (APL; M3)
MPO/SB ++ ; SE +
NSE -
PAS diffuse
(staining patterns of AML) acute myelomonocytic leukemia (M4)
MPO/SB ; SE ; NSE +
PAS diffuse
(staining patterns of AML) acute monocytic leukemia (M5)
MPO/SB +/-
SE -
NSE +
PAS diffuse
(staining patterns of AML) acute erythroid leukemia (M6)
MPO/SB + (blasts)
SE / NSE -
PAS + (rbc precursors)
(staining patterns of AML) acute megakaryocytic leukemia (M7)
MPO/SB ; SE -
NSE + (megakaryoblasts)
PAS +/-
WHO classification of AML
Emphasizes cytogenetic and molecular features in classifying AML
Reduced blast threshold from 30% → 20%
20% = threshold that differentiates an acute leukemia from chronic and MDS
AML treatment/prognosis
Chemotherapy—eradicate all the malignant cells in BM, allowing for repopulation with normal hematopoietic precursors
three groups of agents may be used (antimetabolites, alkylating agents, and antibiotics)
e.g., cytosine arabinoside, daunorubicin, amsacrine
Radiotherapy—prevent or eradicate leukemic cells that have infiltrated the meninges
BM transplant: Highest rate of success in younger (<30 yrs) pts
(acute leukemias) acute lymphoid leukemia (ALL) presentation
symptoms similar to that of AML
lab findings for ALL (wbcs, rbcs, plts)
WBCs:
Increased, normal or decreased
Neutropenia ; Lymphoblasts
RBCs: normochromic, normocytic anemia
Platelets: thrombocytopenia
lab findings for ALL (BM, cytochemical stains)
BM: Hypercellular >20% blasts, usually heavily infiltrated
Cytochemical staining not always effective in determining the cell lineage of undifferentiated cells
several monoclonal antibodies have been identified to help determine the specific classification of ALL
useful markers for subclassification of ALL (chart)
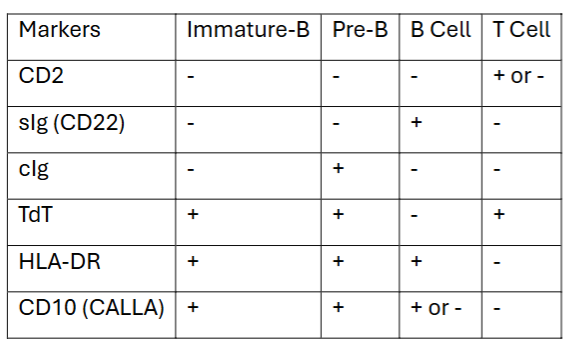
distribution of ALL phenotypes
65% = Immature B cell
20% = Pre-B cell
1% = B cell
15% = T cell
treatment/pronosis of ALL
usually involves three phases: induction (putting the patient into remission), CNS prophylactic phase, maintenance chemotherapy
Chemotherapy: Nearly all children and >90% of adults achieve remission in this manner
>80% of children who achieve remission and a 2–3-year regimen of maintenance therapy are thought to be cured ; adults have only 20–30% long-term survival rate
Intrathecal chemotherapy and cranial irradiation: prevent or eradicate leukemic cells that have infiltrated the meninges of the brain
BM transplant or stem cell transplant
primary cause of death is infection due to the granulocytopenia; bleeding is the second most common complication
(chronic lymphoproliferative disorders) chronic lymphocytic leukemia (CLL) etiology/pathophysiology
malignant monoclonal proliferation and accumulation of lymphocytes
generally B-cells (95%) and are immunologically incompetent cells
primarily disease of the elderly with 90% of patients >50 years; 65% are >60 yrs
occurs twice as often in men as in women
In some rare cases the CLL is the result of T-cell proliferation (2–5% of cases)—more likely to have infiltration of epidermal sites and CNS
(chronic lymphoproliferative disorders) CLL presentation
very insidious onset
fatigue and reduced exercise tolerance → most frequent presenting symptoms
dx often made when the patient presents for investigation of some other problem
enlarged lymph nodes and splenomegaly are common; hepatomegaly develops as the condition progresses
more advanced disease → marked fatigue, bruising, pallor or jaundice with anemia, fever, recurrent or persistent infection, bone tenderness, weight loss, and edema (from lymph node obstruction)
(chronic lymphoproliferative disorders) CLL lab findings
Absolute lymphocyte count 10–150x103/μL common ; can be as high as 1,000x103/μL
Lymphs appear to be morphologically normal—somewhat fragile and easily "smudge" when a blood smear is made
dense chromatin, nucleoli and agranular cytoplasm
Autoimmune hemolysis may account for an anemia in 5–10% of patients and may be triggered by viral infections, disease progression, therapeutic agents, or membrane damage by abnormal proteins
Plasma immunoglobulins may be decreased
BM sample usually not required
(chronic lymphoproliferative disorders) immunological classification of CLL
lymphocytes in B-CLL appear as mature cells and as such are not identifiable using morphology criteria alone
Markers Useful in Identifying B-CLL Cells:
Surface Immunoglobulin (sIg)
almost always express low amounts (one-tenth of normal B-cells)
sometimes demonstrate cIg
CD5 (usually thought of as a T cell marker)
CD19, 20, 22
(chronic lymphoproliferative disorders) differential dx for CLL
Reactive lymphocytosis
Hairy cell leukemia
Sezary's syndrome
T-cell CLL
Prolymphocytic leukemia
Well-differentiated diffuse lymphoma
Poorly differentiated lymphoma
(chronic lymphoproliferative disorders) CLL treatment
chemotherapy: eradicate malignant cells in BM, allowing for repopulation with normal hematopoietic precursors
ex: cyclophosphamide or chlorambucil is given singly or in combination with vincristine and prednisone
Main risk with this treatment is myelosuppression of the bone marrow
Radiotherapy: treat enlarged lymph nodes and splenomegaly resistant to chemotherapy; does not reduce the number of lymphocytes in the blood and marrow
Leukapheresis: reduces number of lymphocytes in blood
(chronic lymphoproliferative disorders) CLL prognosis
clinical course may range from extremely benign to severe with 10—15% of patients living 10–15 years with little or no treatment
death may come within one year for those with a more aggressive form of the disease—median survival following diagnosis is 3–4 years
CLL is not as likely to undergo a transformation to an acute form as are other leukemias
leukemic cells may infiltrate other organs: skin, prostate, gonads, kidney, walls of the gastrointestinal tract
(chronic lymphoproliferative disorders) hairy cell luekemia (HCL) etiology/pathophysiology
distinct clinical entity characterized by pancytopenia, splenomegaly, and presence of a unique cell type ("hair cells") in peripheral blood and tissues and a dry tap bc of marrow fibrosis
Relatively rare (2% of all leukemias)
growth and accumulation of the hairy cells in the spleen, blood and marrow account for the complications the patients experience
men more affected than women (4:1 to 5:1 male: female)
Median age: 50 yrs—not seen in patients <20 years
(chronic lymphoproliferative disorders) hairy cell leukemia presentation
indolent course in most patients
Bleeding, weakness and fatigue, infection and abdominal discomfort
Splenomegaly seen in 80% of patients
Lymphadenopathy and hepatomegaly less frequently seen
(chronic lymphoproliferative disorders) hairy cell luekemia (HCL) lab findings
hairy cells: in peripheral blood of >90% of patients
represent <50% of leukocyte differential count
Scant to abundant, agranular, light grayish-blue cytoplasm with hair-like or ruffled projections
Pancytopenia: Granulocytopenia and monocytopenia; normochromic, normocytic anemia
BM: dry tap due to fibrosis; increased reticulin (fibrosis
Cytochemistry: TRAP staining (characteristic but not diagnostic) in hairy cells but negative in most other lymphocytes
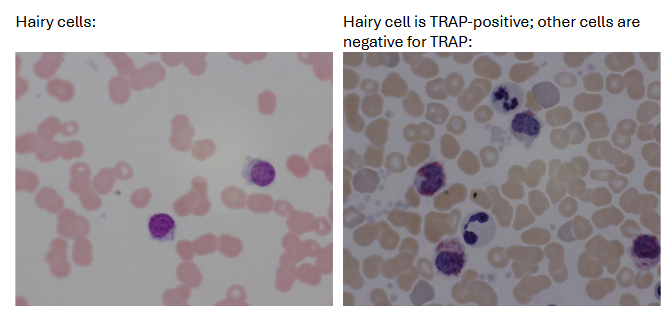
(chronic lymphoproliferative disorders) immunological classification of HCL
immunologically, the hairy cells are a mid- to late B-cell with sIg and cytoplasmic immunoglobulin
useful markers for B-HCL Cells
Negative = CD5
Positive = CD19, 20, 22, 25, 103, 11c
(chronic lymphoproliferative disorders) hairy cell luekemia (HCL) treatment/prognosis
median survival is 5–7 years
malignant lymphoproliferative disorders (list)
Hodgkin's Lymphoma
Non-Hodgkin's Lymphoma
Multiple Myeloma
Plasma Cell Leukemia
Waldenstrom's Macroglobulinemia
(malignant lymphoproliferative disorders) hodgkin’s lymphoma etiology/pathophysiology
Uncertain cause
infectious cofactor appears to be operational in the pathogenesis of the disease; probable agent is EBV; CMV & herpesvirus 6 have been implicated
Incidence:
May occur at any age
Bimodal distribution: 20–30 yrs then, in patients >50 yrs
Most common malignancy of young adults in US and Europe
Accounts for 1/3 of newly diagnosed lymphomas in US
(malignant lymphoproliferative disorders) hodgkin’s lymphoma presentation
Adenopathy and no symptoms, or fever, night sweats, weight loss, malaise
Unusual complaints → pain in enlarged nodes after alcohol consumption
Lymph node involvement:
Most frequent sites are cervical and supraclavicular lymph nodes
Later spreading to contiguous nodal regions, spleen, liver, and BM
Subclassified into 4 types based on histopathology of involved lymph nodes
(malignant lymphoproliferative disorders) hodgkin’s lymphoma lab findings
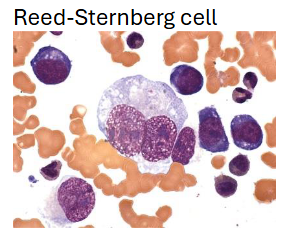
Cytologic hallmark is Reed-Sternberg cell (RS)
Data favors a lymphocyte origin of RS cell–either B/T-cell
Dx based upon finding RS cells in the proper cellular, stromal, and clinical setting
RS-like cells commonly seen in a variety of benign & malignant conditions other than Hodgkin's Disease
Viral infection (ex: mono); anticonvulsant-induced lymphadenopathy; epithelial & stromal malignancies; melanoma; various lymphomas & leukemias; myeloproliferative disorders
(malignant lymphoproliferative disorders) hodgkin’s lymphoma clinical stages (4)
Stage I: Tumor in 1 anatomic region or 2 contiguous anatomic regions on same side of diaphragm
Stage II: Tumor in >2 anatomic regions or 2 noncontiguous regions on same side of diaphragm
Stage III: Tumor on both sides of diaphragm not extending beyond lymph nodes, spleen, or Waldeyer's ring (includes the palatine, pharyngeal and lingual tonsils that encircle the pharynx)
Stage IV: Tumor in BM, lung, etc. (any organ site outside of the lymph nodes, spleen, or Waldeyer's ring)
(malignant lymphoproliferative disorders) non-hodgkin’s lymphoma (NHL) etiology/pathophysiology
Environmental (chemicals, and ionizing radiation, & certain viruses) and inherited genetic abnormalities may participate in the production of irreversible chromosomal alterations that underlie NHL
Incidence: Freq is age dependent (more common in adults than children); variable worldwide distribution; more common in males than females
(malignant lymphoproliferative disorders) non-hodgkin’s lymphoma (NHL) presentation
Can be grouped morphologically into small, intermediate, or large size cells with diffuse or follicular (nodular) pattern of growth
functionally into B-cell, T-cell, and U-cell (undifferentiated cell) disorders
Clinical setting varies with the type of lymphoma
Prognosis generally worsens with increasing cell size and mitotic activity
(malignant lymphoproliferative disorders) non-hodgkin’s lymphoma (NHL) lab findings
Demonstration of surface and/or intracytoplasmic markers of lymphocytes extremely valuable in the classification
Marker studies can determine the T- or B-cell nature of the cells—utilize flow cytometry and immunoperoxidase techniques
(malignant lymphoproliferative disorders) multiple myeloma etiology/pathophysiology
Uncontrolled proliferation of malignant plasma cells in BM
Incidence: Most common form of plasma cell neoplasm
Males have 50% greater risk than females
Black individuals have 2x incidence than white individuals
Risk increases with age
Environmental factors play clear role for instance, atomic bomb survivors & individuals exposed to radiation have increased risk
(malignant lymphoproliferative disorders) multiple myeloma presentation
Multiple osteolytic bone lesions
Tumors develop in hemopoietically active marrow, erode adjacent bone, and may compress the spinal cord or nerve roots emerging from the spinal canal
Skeletal disease seen in 70% at diagnosis
Imbalanced immunoglobulin production most frequently yields an excess of light chains that are excreted into urine
(malignant lymphoproliferative disorders) multiple myeloma lab findings
Monoclonal gammopathy – Incidence: IgG>IgA>IgM>IgD>IgE
Generalized hypogammaglobulinemia with Bence Jones proteinuria
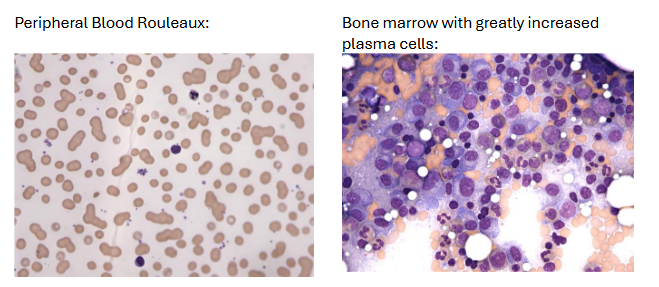
Peripheral blood: Rouleaux, increased ESR
Bone marrow: Varying number of mature and immature plasma cells
Cells are frequently enlarged with less chromatin clumping than seen in benign plasma cells
Multinucleated cells seen
(malignant lymphoproliferative disorders) plasma cell leukemia
when plasma cells predominate among the circulating leukocytes it is known as leukemia instead of multiple myeloma
protein abnormalities similar to classic myeloma
clinical course is generally acute or sub-acute
No response to chemotherapy observed
(malignant lymphoproliferative disorders) waldenstrom’s macroglobulinemia
plasma cell dyscrasia that secretes monoclonal IgM
differs from multiple myeloma instead of osteolytic bone lesions there is soft tissue involvement
lipid storage diseases (general)
hereditary conditions in which the macrophages are unable to completely digest phagocytosed material due to a deficiency of lysosomal enzymes needed for the degradation process—undigested substance accumulates in the cell; includes:
Gaucher & Niemann-Pick Disease
Sea-blue Histiocytosis Syndrome
Tay-Sachs & Sandhoff Disease
Fabry & Wolman's Disease
(lipid storage diseases) gaucher disease etiology/pathophysiology
Deficiency of B-glucocerebrosidase
Macrophage unable to digest stroma of ingested cells & glucocerebroside accumulates in cell
Incidence: autosomal recessive; predominantly in Jewish populations, esp Ashkenazic Jews
(lipid storage diseases) gaucher disease presentation
Related to accumulation of the lipid in macrophages of lymphoid tissue, spleen, liver, and BM
Hepatosplenomegaly
Neurological problems
Pigmentation of skin (ocher to brown with a yellow or leaden hue); prominent in exposed parts of the body (face, neck, hands, legs)
Skeletal lesions secondary to marrow involvement; bone pain
(lipid storage diseases) type 1 gaucher disease
adult, chronic non-neurotologic
Most common of the lipidoses
Most frequently inherited disorder in Ashkenazi Jewish population
Average age of onset is between 30–40 yrs; best prognosis
(lipid storage diseases) type 2 gaucher disease
infantile, acute or malignant neuronopathic type
Very rare; seen in all ethnic groups; uncommon in Jewish population
Familial intermarriage is frequent in infant's family history
hallmark is neurologic involvement
Death usually occurs before age of 2 yrs
(lipid storage diseases) type 3 gaucher disease
juvenile type; subacute neuronopathic
Onset of symptoms between early childhood to teenage years
Clinical & physical findings and survivals range between those of Type I and II
Noted especially in group of children from northern Sweden (offspring of several related intermarriages)
(lipid storage diseases) gaucher disease lab findings
Peripheral blood: (result from cellular sequestration by an enlarged spleen)
Anemia: Normochromic/normocytic
Retics: Normal to modest increase
WBC: Mild leukopenia
Gaucher cells
Platelets: Slight to marked decrease
Serum acid phosphatase: Increased
Mild extravascular hemolysis
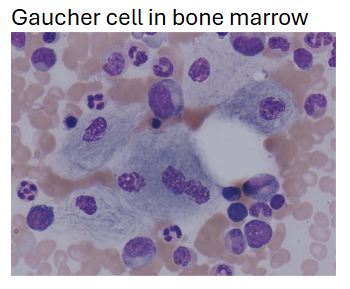
(gaucher-disease) guacher cells
Found in spleen, marrow, liver, lymph nodes; seen most easily in thickest part of the smear
Cytoplasm appears wrinkled liked crumpled tissue paper
BM exam alone insufficient for confirming diagnosis since the cells may be present in other disorders
Cytochemistry: PAS, Oil Red O, Iron, and Sudan Black B are positive; Acid Phosphatase is very positive (tartrate-resistant fraction is what is increased)
psuedo-gaucher cells
found in patients with high cell turnover where the breakdown products cannot be
catalyzed fast enough
Described in AML, CML, plasma cell myeloma, aplastic anemia, ITP, thalassemia major,
and RAIn each of these diseases there is no deficiency of the b-glucocerebrosidase, as there is in Gaucher disease, but rather an over taxation of a normal system
(lipid storage diseases) gaucher disease treatment/prognosis
enzyme replacement therapy has successfully reversed many of the clinical complications of Type I disorder, including correcting blood counts & reducing organomegaly
(lipid storage diseases) niemann-pick disease etiology/pathophysiology
Deficiency of sphingomyelinase
Secondary accumulation of unmetabolized lipid sphingomyelin as well as cholesterol
Incidence: rare autosomal recessive; more commonly seen in Ashkenazic Jewish population
5 types (A - E) based on clinical manifestations
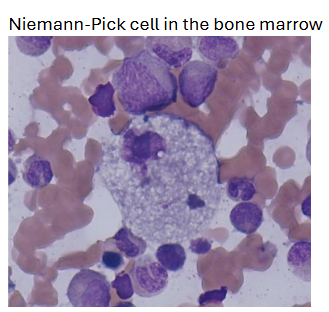
(lipid storage diseases) niemann-pick disease lab findings
leukopenia and thrombocytopenia may occur
(lipid storage diseases) niemann-pick disease treatment/prognosis
no treatment ; successful allogeneic BM transplants have been reported for type B