Basics of Pharmacology (Lectures 1-3)
1/87
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
88 Terms
pharmacology
study of the effects of chemical substances on living systems through chemical processes (activating or inhibiting) to produce beneficial therapeutic effect
bind to regulatory molecules, modification of receptor
how does pharmacology differ from pharmacy
pharmacy uses the principles learned from pharmacology in clinical settings; mainly in a dispensing or clinical care role
when was pharmacology named
~200 - 250 years ago
early stages of pharmacology was:
herbal remedies >3000 years ago
earliest: important plants by indian and egyptian scholars (15th and 6th century BC)
who played major roles in prevalent clinical practices
religious authorities
limitations of crude plant-derived materials
outcome was uncertain
knowledge of normal and abnormal functioning of the body was very limited
disease and death was semi-sacred subjects
other prevalent remedies
not based on rationale principles (limited use)
allopathy - emetics and purgatives (effectiveness not clear)
homeopathy - guided principles like cures, and the effect of a drug is enhanced by dilution (more dilution = more potent?)
3 main pillars forming the foundation of modern pharmacology
physiology, pathology, chemistry
cell theory from 1858
all living organisms are composed of one or more cells
cell is the basic unit of structure and organization of organisms
cells arise form pre-existing cells
major events of 1868 and 1878
first description of strucutral formula and synthesis of an organic compound (urea)
discovery of bacteria as the cause of disease by Pasteur
progress of biochemistry 20th century
discoveries of enzymes and chemical pathways - provided the framework for understanding the precise effect of various drugs
biotech and genomic era
mid 1980 to present
use of biotech for the production of antibodies, enzymes, growth factors, hormones, gene therapy ex. insulin
previously needed to purify, limited product, extensive process before obtaining pure
controlled clinical trials
use of a control group (receive either placebo, another treatment, no treatment) in clinical studies
introduced 70 years ago
allows evaluation of clinical effectiveness of any substance
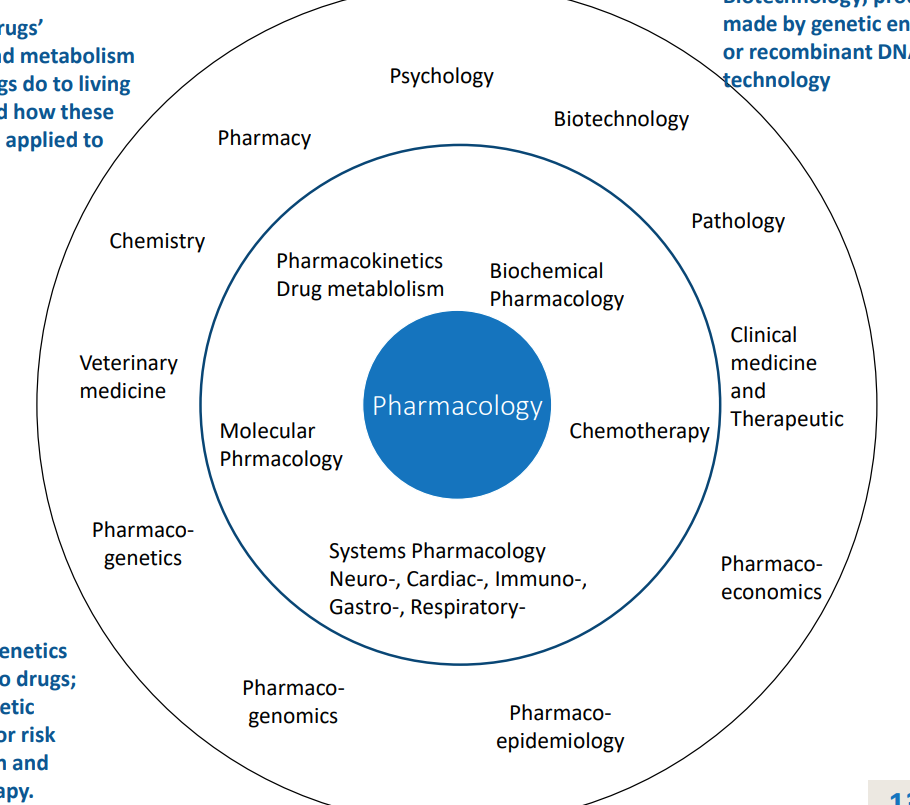
core areas of pharmacology
drug absorption, metabolism, effect on living organisms, how effects can be applied to therapeutics
define biotechnology
products made by genetic engineering or recombinant DNA technologies
pharmacology wheel
Drug dosage
must be adjusted to each individual need
define a drug
any substance causing change in biological function through chemical interactions that is therpeutically useful
examples of solid, liquid, gas drugs
aspirin
nicotine, ethanol
nitrous oxide (at room temp)
what do physical natures of drug determine
route of administration, therapeutic effect, rate of absorption, distribution, and elimination in the body
cascade initiated once a drug is given
how can drugs be synthesized
hormones can be synthesized in the body
produced outside the body synthetically
how do most drugs work
interact with a specific receptor molecules and alter the receptor’s biochemical or biophysical activities
2 categories of drug body interaction
pharmacokinetics and pharmacodynamics
pharmacokinetics
what body does to the drug (absorption. distribution, metabolism and elimination)
pharmacodynamics
actions of the drug on the body (different chemical processes to bring about effect)
range of molecular drug size
7Da to 59050Da, but majority 100-1000Da
importance of the lower limit of a drug
for the need of specificity, should be large enough to be unique in shape, charge, etc to prevent non-specific binding to other receptors
importance of the upper limit of a drug
determined by the ability of molecules to diffuse between compartments of the body, large drugs will not diffuse readily in body compartments - often administered directly to the compartments in which they exert their effect
aim of drug therapy (pharmacokinetics)
prevent, control, or cure diseases
requires delivery of adequate (therapeutic) doses of the drug to the target tissue at levels that are generally non-toxic
consideration of barriers such as lipid membranes, drug elimination
determines the concentration of a drug at its sites of action
barriers within mechanism of drugs absorption, distribution, permeation
lipid membranes, drug elimination
knowledge of pharmacokinetics allows for
prediction of route of administration, doses, dosage regimens
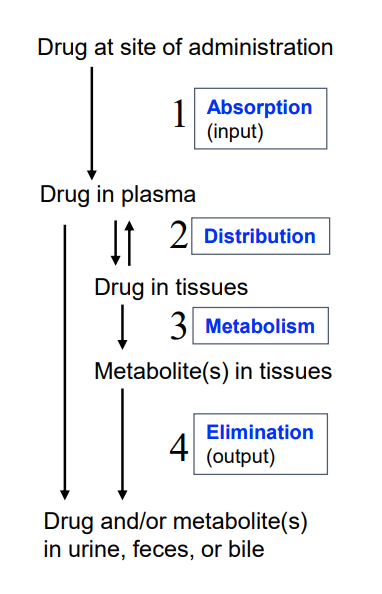
Drug movement in the body
site of administration
plasma (distribution)
tissue (metabolism)
output (elimination)
2 main categories of routes of drug administration
enteral and parenteral
enteral administration
all or large portion of the drug passes through the gastrointestial tract (GI)
oral, rectal, sublingual
parenteral administration
GI tract mostly avoided
intravenous, intramuscular, subcutaneous
other routes of drug administration
inhalation, topical, intranasal, transdermal
enteral route
mouth, esophagus, stomach, small intestine, anus
oral administration
most common, amount of drug that enters the system is affected by its absorption and by the drug metabolism in the liver
sublingual administration
under the tongue, absorbed through capillaries, >50% of drug bypasses first-pass metabolism in liver, 50% passes to GI tract
nitroglycerin >90% inactivated by first pass metabolism
rectal administration
absorption through blood vessels in the rectum, >50% bypasses the liver, metabolism is minimized
useful in cases where drug is inactivated by acidic pH of the stomach or intestinal enzymes
also for drugs that induce vomiting or if patient is already vomiting
commonly used for antiemetic drugs, allows for administration outside a clinical setting
reasons for parenteral administration
drugs can be unstable, inactivated or metabolism by GI tract, poorly absorbed by oral administration, could be charged molecule or hydrophobic, rapid effect
intravenous administration
100% reaches the bloodstream
most control over doses, generally administered in hospital setting
if administered by IV it cannot be easily reversed by emesis (vomiting)
must be administered slowly, concentration cannot be too high
can also lead to bacterial contamination or infection inadvertently
will eventually be inactivated in the blood, but will take time
intramuscular administration
some in this form given in depot form (suspension in non-aqueous solvent like polyethylene glycol)
as solvent diffuses, drug dissolves at site of injection, providing a sustained release of the drug for extended duration (drug will dissolve slowly, longer term effects)
drug effective once it has dissolved, therefore concentration at the site is not an issue
mainly vaccines and single injection of drugs
subcutaneous administration
requires absorption and is slower than IV
contraceptive capsules for long term activity, can be implanted
allows embedment of programmable devices (mechanical pumps to deliver insulin to diabetic patients)
subcutaneous vs intramuscular injection
difference in angle of injection
inhalation administration
absorption through mucous membranes
drugs in gaseous form (anesthetics) or an aerosol
useful in respiratory conditions (asthma), drug delivered right to site of action (not system wide)
topical administration
application on the skin (ointment, cream)
not system wide effect (local)
first pass metabolism
in the liver (and often kidneys) before entering circulation via absorption from stomach and duodenum
can greatly limit the system availability of the drug
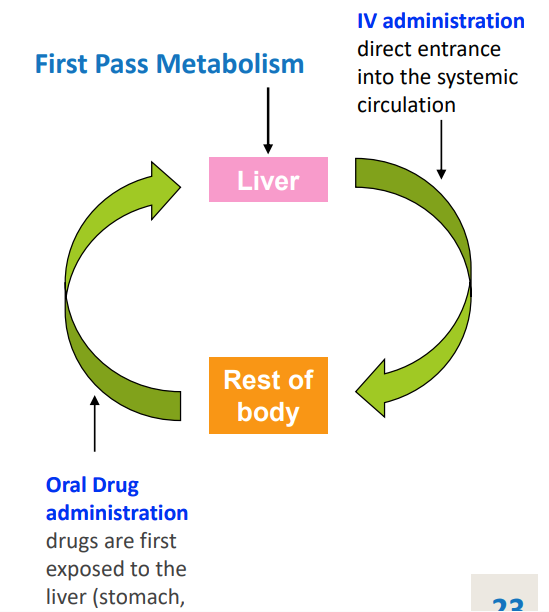
how much of the drug will bypass the liver when deliveryed by rectal or sublingual
50%
what increases the duration of a drug in the stomach
presence of food (can also affect metabolism and absorption) ex. penicillin inactivated by acid
what is absorption
transfer of a drug from its site of administration to the bloodstream (dependent on route of administration)
absorption via intravenous delivery
intravenous has close to 100% bioavailability and doesn’t have to be absorbed in the same manner as other routes of administration because all given dose reaches the systemic circulation
what is bioavailability
fraction of the administered drug that reaches the systemic circulation from where it has access to the site of its action
most important mechanism for drug absorption
passive diffusion across a lipid bilayer
various aqueous body compartments are separated from each other by lipid barriers
most drugs will have to cross multiple anatomical barriers before reaching their sites of action
where are drugs taken orally absorbed to produce an affect on nervous system
walls of the intestine
walls of the capillary that perfuse the gut
blood brain barrier
walls of the capillaries that perfuse the brain
components of a lipid bilayer
polar heads, hydrophobic tails
aqueous parition coefficient
determines how easily drugs move between aqueous and lipid media
lipid diffusion depends on:
soluability of the drug
charge of most drugs
weak acids or weak bases (charged states and lipid soluability varies with the pH of the medium)
passive diffusion
driven by a concentration gradient (in the body, contains larger aqueous compartments)
what compounds will not allow drugs to pass through the basal membrane
binding to larger proteins, limiting factor
pores in capillary cell walls permit passage of molecules as large as 20-30 kDa
what law defines passive flux across a concentration gradient
Fick’s Law
Flux equation
(C1-C2)*(area x permeability coefficient)/thickness)
C1: higher concentration
C2: lower concentration
Area: area across which diffusion is occuring
permability coefficient: measure of mobility of drug in particular medium
thickness: length of diffusion path
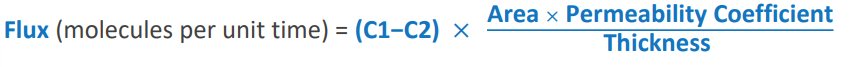
effect of pH on drug absorption
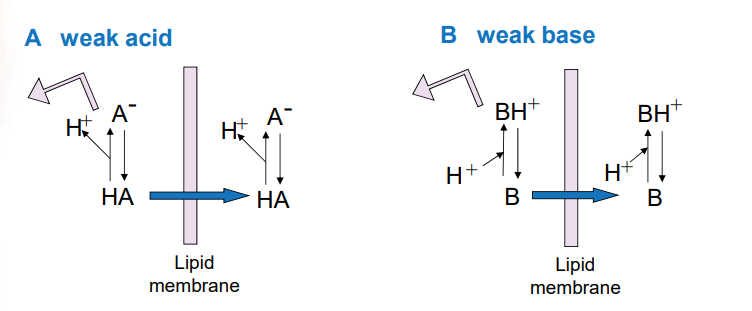
uncharged form of the drug passes through the membrane more readily, relative concentration of the 2 forms is dependent upon pH at the site of absorption and pKA of the acid or base
weak acid: binds to H+ to be neutral and passes through
weak base: gives up its H+ to be neutral and passes through
henderson-hasselback equation
ratio of protonated to unprotonated molecules of a drug with certain pKA, in a medium of given pH

more weakly acidic drugs will be in lipid soluable form at ____ pHs
acidic
more weakly basic drugs will be in lipid soluable form at ___ pHs
basic
when pH is less than pKa, the ______ forms of ___ and ___ predominate
protonated forms of HA and BH+ predominate (ionized form, can be trapped)
more H+ in the environment = more HA and BH+ protonated forms
when pH is greater than pKA, the _____ forms of ___ and ___ predominate
unprotonated forms A- and B predominate (unionized form, won’t be trapped)
less H+ in the environment means more A- and B unprotonated form, therefore easier to transport and absorb
when pH = pKA,
HA = A- and BH+ = B
the unionized form will be the same on both sides of the membrane
pHs of the 2 compartments will determine how much total drug will be on each side
the compartment where the degree of ionization is
what may cause trapping of a drug into certain compartments
pH of some bodily fluids; different from the blood
will be due to a greater concentration of the drug due to its ionization
effect of pH on drug distribution in body compartments
aspirin example of pH dependence
aspirin is highly unionized in the gastric juices
accumulates to high concentrations inside the cells of the stomach mucous membrane (have close to neutral pH)
leads to adverse effects as gastritis or gastric ulcers because in the mucous membrane it ioninzes and cannot extract
increases concentration of aspirin in the stomach
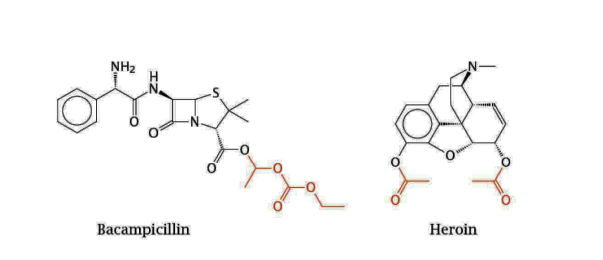
how can you increase absorption of ionic drugs
mask the charged groups
ex. heroin is morphine + 2 ester groups which are cleaved upon entry into the body by esterases, and releases morphine
ex. the ester groups of bacampicillin can be cleaved to release ampicillin after uptake into the intestinal epithelium
what is carrier-mediated membrane transport
active and facilitated transport including molecules that can transport drugs across a membrane
active is dependent upon metabolic energy
what types of drugs are transported via carrier-mediated membrane transport
molecules that have similar structures to peptides, nucleosides, nucleotides, and are related to natural substrates
acting on central nervous system (e.g. L-dopa, L-a-methyldopa, GABA), and oral beta-lactams (amoxicillin, cephalexin)
2 types of carrier mediated transports
active and facilitated (only active is dependent on metabolic energy)
define active transport
energy dependent and is driven by the hydrolysis of ATP (or by ion gradient) capable of drug transport against a concentration gradient
Such carriers are saturable (reaches maximum velocity at high substrate levels) and inhibited in the same manner as enzyme-catalyzed reactions
characteristics of facilitated diffusion
Requires a carrier to mediate transport of drugs/molecules that will diffuse too slowly through membrane. Examples; different types of ion channels
Does not require energy input
Cannot occur against a concentration gradient. Shows greater temperature dependency
endocytosis/pinocytosis of drugs
Mechanism used for absorption of drugs
100 kDa such as proteins, toxins, antigens etc., Uptake of vitamin B12, complexed with a binding protein, across the gut wall; Uptake of iron bound to the protein transferrin by hemoglobin synthesizing cells
Molecules are engulfed by cell membrane and carried into the cell by pinching off of the newly formed vesicle;
Exocytosis: reverse of endocytosis- e.g. release of neurotransmitters
what are the physical factors influencing drug absorption
blood flow to the absorption site - Blood flow to the intestine is much greater than to the stomach, so absorption from the intestine is favored
total surface area available for absorption - Generally the rate and quantity of drug absorbed is directly proportional to area available for absorption. Intestine surface is rich in villi and microvilli increasing its surface area about 1000 fold more than that of stomach
contact time at the absorption surface - Greater contact time = more absorption
If a drug moves through the GI tract rapidly as in diarrhea, it is not well absorbed
anything that delays the transport of the drug from stomach to the
intestine (food in the stomach) delays the rate of absorption of the drug or its entrance into the general circulation.
general pathway of drug absorption
passive diffusion across the epithelial cell layer lining the intestine and from there to blood capillaries (another single cell layer); unabsorbed drugs or nutrients are excreted in feces
Blood capillaries drain their contents into hepatic portal veins leading to liver
From liver the blood goes to general circulation in the body
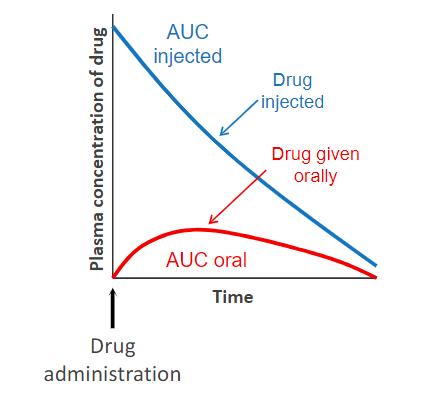
determination fo bioavailability
Comparing plasma levels of a drug after a particular route of administration with
plasma drug levels achieved by IV injection (of the same drug dosage), in which all of the given drug enters the circulation
factors that influence bioavailability of orally administered drugs
Absorption from the Gut
Solubility of drug: very hydrophilic drugs poorly absorbed; extremely hydrophobic drugs insoluble in aqueous body fluids.
Chemical instability
Nature of the drug formulation e.g. particle size, binders that can affect
its dissolution.First pass Metabolism (extraction ratio)
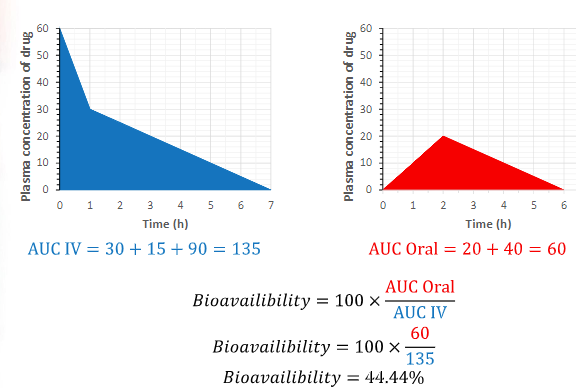
bioavailability equation
100 x (AUC Oral / AUC IV)
what is bioequivalence
Two related drugs which show comparable bioavailability and similar times to achieve peak blood concentrations
what is therapeutic equivalence
Two similar drugs which have comparable efficacy and safety