PMCOL 306 Administration, Absorption, Bioavailability
1/89
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
90 Terms
What is intra-arterial administration?
injection of drug into an artery to localize its effect in an organ
What is the definition of dose?
amount of drug administered (mols)
What is the definition of volume of distribution?
volume which a drug is distributed (L)
What is the definition of Concentration
Amount of drug in a give volume plasma (M)
What is the definition of absorption rate constant (Ka)?
rate at which a drug enters the body (h^-1)
What is the definition of elimination half-life (t1/2)?
time it takes for the concentration of the drug to reach half the original value (h)
What is the definition of elimination rate constant? (k, ke, kel)
rate at which drug is removed from body (h^-1)
What is the definition of infusion rate? (kin)
rate of infusion required to balance elimination (mol/h)
What is the definition of AUC?
area under the curve; integral of the concentration-time curve
What is the definition of clearance? (CL)
volume of plasma cleared of the drug per unit time (L/h)
What is the definition of bioavailability? (f)
fraction of drug that is systemically available
What is the definition of Bolus?
Single dose administered (usually intravascular).
What is the definition of Infusion?
Continuous administration
What is the definition of Extravascular and Intravascular?
Any route of administration besides intravascular (absorption rate and bioavailability become important factors) — Extravascular
Direct administration to blood pool (instantaneous absorption) — Intravascular
What is the definition of Dosing interval (Tau, τ)
Time between drug dose administrations (h)
What is the definition Cmax, Tmax, and Cmin
Cmax — The peak plasma concentration of a drug after administration. (M)
Tmax — Time to reach Cmax.(h)
Cmin — The lowest (trough) concentration that a drug reaches prior to the next dose. (M)
What is the definition of Fluctuation (%PTF)
The ratio between Peak (Cmax) : trough (Cmin), within one dosing interval at steady state. (%)
What is Enteral vs. parenteral?
enteral - GI absorption
parenteral - everything else
What 3 administration routes are enteral and rank them in terms of onset effect?
oral (lowest) --> rectal --> sublingual (fastest)
What qualities effect the choice of administration? (5)
physical drug propertie
(solid, liquid and gas)
chemical drug properties
(solubility, stability, pH, logP [lipid solubility], irritancy)
site of desired action
(local or generalized)
effect of digestion and first-pass metabolism
Effects of digestive juices, extent of first pass metabolism, extent of absorption
condition of patient
What are the 4 different routes of administration from the skin
Transdermal (just below top layer of the skin)
Intradermal (blow the skin in the dermis lay (2nd from the top))
Subcutaneous (into fat)
Intramuscular (into muscle)
What are some of the challenges of transdermal administration?
Hard from most drugs to pass through the outer most layer of skin — the stratum corneum
stratum corneum is comprised of keratin-filled corneocytes which are anchored in a lipid matrix. This makes a brick and mortar making it hard from chemicals to pass through
How does a drug get absorbed when administered transdermally (3)? And what solutes would use each pathway (think chemical characteristics)
intracellular route
hydrophilic or polar solutes
intercellular route
lipophilic or non-polar solutes
transappendegeal route (passage of molecules through sweat glands and hair follicles)
high molecular weight molecules, charge molecules
What are some of the pros and cons of transdermal absorption
Pros
Painless, non-invasive, easy to use
High bioavailability (bypass first pass metabolism)
Cons
inefficient absorption (~1-2%), absorption depends of surface area and high logP
Would high lipophilicity increase or decrease transdermal adsorption?
increase
What are ways to enhance transdermal absorption? (3)
- abraded or burned skin (removed layers)
- suspended drug in oil to increase lipophilicity (oily vehicle)
- ion pairing (make neutral complex that can breakdown once in aqueous environment)
Intradermal Injection
Drugs that would administered this way
speed comparisons with subcutaneous
Con
Would be used to administer drugs that are unable to bypass the stratum corneum because of they are less lipophilic
Faster onset affect (close to blood vesicles)
Con - high skill need to hit the right layer
Subcutaneous
Drugs that would administered this way
Why a drug would be administered this way
Use (rate of absorption)
Cons (2)
Pro
non-ionic and high lipid solubility, non- irritant (in fat)
drugs that have poor GI absorption are injected subcutaneously
slow and constant rate of absorption (slow to leave fat cells and low blood flow to fat) cause sustained effect. (effect may be desirable — could implant solid pellet form of drug in fat — effect over months)
Con
Drug can’t be an irritant causes pain, necrosis, inflammation
Pro
cheep infusion compared to IV
Don’t need to me monitored my medical personal
Intramuscular
Rate of absorption (2)
Ways to influence absorption
Sites of injection (good and bad)
faster rate of absorption than subcutaneous or intradermal injections (because muscle is well vascularized)
Oily drugs = slow constant absorption — aqueous drugs fast rapid absorption
thinks ways to increase blood flow — Temperature, exercise, massage
Site of injections
Dorsolateral (back of the butt)
Bad— lots of nerves and important blood vessels
Ventrogluteal (side of butt) — the best
Better because less likely to hit the important stuff
Women absorb in the butt slower because they have for fat there
Deltoid — the most common
Ideal place — accessible, faster than butt injections
Are injection administration routes subject to first-pass metabolism?
no
Sublingual (SL— under the tongue) and Buccal (cheek) administration
Type of drugs
Mechanism of absorption
Consideration
non-ionic and highly lipid soluble (most drugs can’t due to erratic absorption)
absorbs in the month into the superior vena cava, by passing systemic circulation
Eating, drinking, smoking can affect absorption
What are the advantages of rectal administration? (6)
- used in cases of nausea, vomiting, inability to swallow
- drugs that taste bad for kids
- rapid systemic effects
- absorption rate not affected by food
- can be easily removed/terminated upon adverse reactions
- some first-pass metabolism avoided (~50% not affected my first pass metabolism)
What are the disadvantages of rectal administration? (4)
- defecation can interrupt absorption process
- absorption can be highly irregular/incomplete
- reduced SA may limit drug absorption, ; low rectal fluids
- patient adherence
What is intrathecal administration?
direct injection into the brain or spinal subarachnoid space (bypass the BBB)
Can drugs administered intrathecally contain preservatives?
no; cannot contain ANY preservatives or potentially harmful ingredients
What are the advantages of pulmonary (lungs) absorption?
- almost instantaneous absorption of drug into blood (large SA in lungs)
- avoids first-pass metabolism
-Local appilcation of the drug
What are the advantages of intravenous administration?
- bioavailability is 100%
- drug delivery is accurate, controlled, and immediate (through titration)
- irritating drugs can be given (slowly bc of dilution)
- only way to administer high molecular weight drugs
-Can either give slow infusion or bolus injections
What type of drug must NEVER be administered intravenously?
What are some other disadvantages
drugs in oily vehicles (cause blood precipitates)
Increase risk of adverse effect — once the drug is injected can’t retreat
What is intra-arterial route of administration
Drug is injected directly into an artery to localize its effect in a particular tissue or organ
What are the advantages of intra-arterial administration?
- greater concentration of drug delivered to desired site
- chemotherapy drugs administered this way decrease systemic toxicity
What are the disadvantages of intra-arterial administration? (3)
- pharmacokinetics vary wildly
- dosages are different than intravenous administratoin
- technically challenging
Need diagnostic agents for this route
What are the advantages of oral administration?
- pre-determined doses, portability, self-administration
- high patient compliance
- most convenient
What is the time of onset for oral administration?
30-90mins
When is absorption favoured for an oral drug?
when drug is in its non-ionized (more lipophilic) form
What are the disadvantages of oral administration?
- high first-pass metabolism
- drug must be stable in acidic environment
What is Lipinski's Rule of Five?
an orally active drug must not violate more than one:
- no more than 5 hydrogen bond donors (total number of N-H and O-H bonds)
- no more than 10 hydrogen bond acceptors (all N or O atoms)
- a molecular mass less than 500 daltons
- octanol-water partition coefficient (log P) that does not exceed 5
What does Lipinski's Rule of Five NOT predict?
it does no predict if a compound is pharmacologically active
Candidate drugs that conform to the Rule of Five tend to have lower what?
attrition rates (less fail) during trials
What percentage of orally administered drugs follow the Rule of Five?
50%
What does the Rule of Five assume?
assumes drugs are absorbed from the gut by passive diffusion (ignores membrane transporters)
What are some factors that affect absorption after oral admin? (6)
ionization (pH) and lipid solubility
Particle size and formulation
Splanchnic Blood flow
microbiota
enzymes (CYP3D4)
efflux pumps
What are the two major transporter classes?
ATP-binding casette (ABC) (efflux transporters) and solute carriers (SLC) (uptake transporters)
What does a negative logP value mean? Positive?
LogP = log10 (partition coefficient)
partition coefficient = [organic]/[aqueous]
negative - compound has higher affinity for aqueous phase
positive - compound has higher affinity for lipid phase
What is a consideration of having a drug with a high logP (highly lipophilic)
The drug will then intern have a low aqueous solubility which would compromise its bootability (won’t from into the blood).
The drug might be suck in fatty tissue and have toxic affects can occur
What logP value is ideal for dugs for the CNS, oral/intestinal absorption, sub-lingual
CNS = 2
oral/intestinal absorption, = 1.35-1.8
sub-lingual = <5
When do drugs that are weak acids become ionized? Weak bases?
weak acid drugs ionize as pH increases (absorbed best in acidic environment)
weak base drugs ionize as pH decreases (absorbed best in basic environment)
Do non-ionized drugs or ionized drugs cross membranes?
non-ionized
How does the pH and pKa affect the absorption of a drug
pH and pKa can affect drug absorption because the ionization state can affect whether a drug is absorbed
How do you calculate ionization ratio (base and acid)
pH that is given is likely to be an area of a body
Acid
pka = pH + log [un-ionized]/[ionized]
Base
pka = pH + log [ionized]/[un-ionized]
Look for the amount of drug drug that is in ionization form (if large amount of non-ionization form = good absorption)
What is oral bioavailability a product of
fraction absorbed (Fa)
Fraction escaping the gut wall (Fg)
Fraction escaping gut wall (Fh)
How do you calculate bioavailability
F= AUCPO (oral admin) / AUCIV
How do you calculate AUCt-∞
AUCt-∞ = Ct (Concentration)/ k (elimination rate constant)
Where in the body do you take a drug measurement from?
blood plasma
What percentage of the human body is water?
50-70%
What percentage of the human body is fat?
20%
What can drugs interact with in the blood
Drugs interact with protein, which prevents it from distributing to other compartments
What are 3 critical factors of drug distribution?
1) concentration gradient of free drug between blood and target organ
2) solubility of drug
3) blood flow to the target organ
Which 3 main compartments do drugs distribute into?
viscera (highly perfused), muscle (well perfused), fat (high lipid soluble drugs)
What are some barriers to drug distribution (6)
pH
Lipid partitioning
Facilitative diffusion and active transporter
Protein binds (either free form (active) or bond form)
BBB
Fat sequestration (getting suck in fat)
What are some details about protein binds and drug distribution
Effect of plasma protein in drug
How to overcome
example
Factors (3)
Only the unbounded drug have any pharmacological effect
Drugs that have extensive plasma protein binding will need a higher dose before having a therapeutic effect
warfarin — 97% bound by protein, 3% is unbound
Factors that impact
Concentration of drug in the body (more drug = more unbound)
Amount of plasma protein (liver disease, renal decrease plasma protein)
Drug-Drug interactions
competitive binding for plasma protein
Drugs and the BBB
Why is it hard for drugs to enter (2)
Hod do you calculate BBB permeability
Methods of entering the BBB
BBB have endothelial cells that are form by tight junction which limit the amount that can enter the brain
basement membrane, pericytes, also contribute to the barrier
logBB = AUCbrain/ AUCplasma
Ways to enter
Paracellular aqueous transport
Transcellular
Efflux pump blocker
receptors mediated transport
magnetitic field mediated
BBB distribution
What plasma proteins commonly bind to drugs?
albumin, alpha-1-acid glycoprotein, lipoproteins
What drugs bind to albumin? What drugs bind to alpha-1-glycoprotein? Lipoprotein?
acidic drugs - albumin
basic drugs - alpha-1-glycoprotein
after albumin and alpha-1-gP are bound --> lipoprotein
If a drug is evenly distributed throughout the body, the _________ will be equal to the ___________.
AVD (apparent volume of distribution), total body water
If a drug has a low AVD, where is majority of the drug?
plasma
If a drug has a AVD around 40 L, what does that mean?
about evenly distributed (lipophilic enough to enter cells but hydrophilic enough to remain in plasma)
If AVD exceeds 40L, what does that mean?
drug has concentrated into tissues
What is ka, ke, km, t1/2, k12, k21, k10?
ka - rate of absorption
ke - rate of elimination
km - rate of metabolism
t1/2 - drug half-life
k12 - distribution from central to peripheral
k21 - distribution from peripheral to central
K110- elimination of central compounds
What does a one compartment model of pharmacokinetic assume?
- drug distributed everywhere
- drug instantaneously and rapidly throughout the body
What is the difference between zero order and first order elimination kinetics?
zero order is a constant amount/time
first order is a constant fraction/time (most common)
What is Vc, Vss, Vβ
VC (volume of central compartment) — The apparent volume of distribution immediately after a bolus (after injected)
Vss (volume of distribution at steady state) — (the gradual increase of Vd)
Vβ (terminal elimination phase half-life) — what causes the constant line
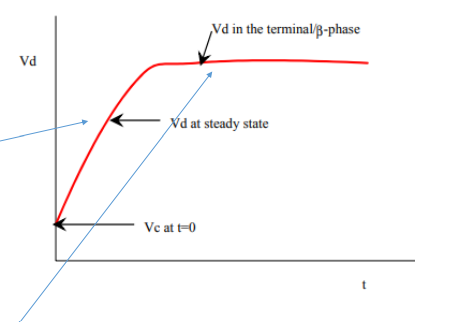
What is the average amount of water in a man and woman
woman — 0.5 L/kg
Man — 0.6 L/kg
What can you tell when
Vc < 3L
Vc ~17L
Vc ~ 40L
Vc >40L
Drug is unable to cross membrane
When drugs bind to plasma more than tissue
Vc < 3L
Drug is mostly in the plasma (heparin)
Vc ~17L
Drug is mostly in extracellular water (mannitol)
Vc ~ 40L
drug is distributed in the whole body (ethanol)
Vc >40L
tissue binding exceeds plasma protein binding (chloroquine)
Drug is unable to cross membrane
that means the Vc cannot be greater the extracellular space
When drugs bind to plasma more than tissue
Vc would be smaller than 40L
What are some factors that affect AVD (other than drugs)
Kidney or liver dysfunction
Would have an increase AVD with liver and kidney dysfunction because plasma binding is lowered
Dehydration
Decrease water = decreased AVD
Obesity
They would have a longer AVD. Because of the higher amount of fat cells highly lipophilic drugs would have longer half-lives and have to consider drug accumulation
Age
Body fat increases and total body water decreases as we age. increase their elimination half life
Why is most drug data ln?
Most drug data shows a exponential decay curve. Which is why the data is most likely to be curved.
How do you calculate one-compartment IV bolus administration
DB=VDCP
Why is estimation of elimination rate constant and half-life are important
the time to reach steady-state
the lime required to eliminate all or a portion of drug from the body
the amount of drug accumulated in the body within a given dosing i interval
the appropriate dosing interval
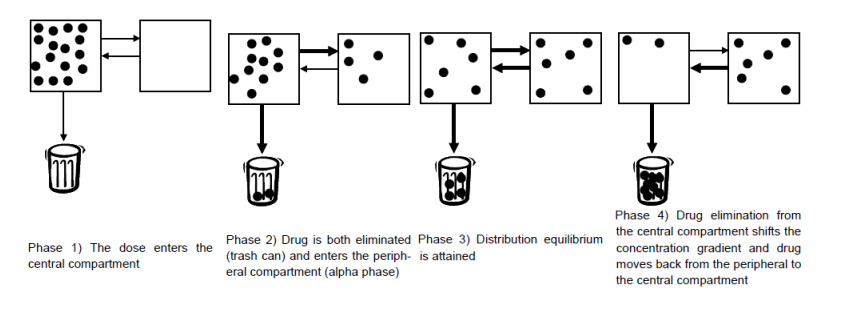
IV - bolus administration (two compartments) describe how the dose travels through the body. (4 phases)
1) the drug in given and it stays in the concentration
2) drug diffuses to the other less perfused tissue and is eliminated from the central compartment
3) the volume of the two reaches EQ
4) The central concentration is less that the less perfused tissue be.
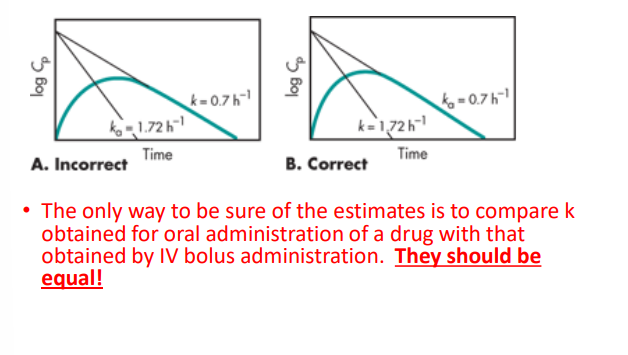
What is flip-flop kinetics
When estimating the oral administration it is assumed that ka > > > > than k.
If K > > > ka than you would fine the Ka and K to be flipped
What is the importance of AI and ADME
Drug development cost a lot of money and not all drugs are able to make it to market
Machine AI is being used to screen for potential toxicity, poor pharmacokinetic properties, biological activity (receptor affinity)