BIOL 2030: Module 11
1/68
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
69 Terms
Type I Restriction Endonucleases
discovered in 1960s
originally thought to be rare, but now found to be very common
not very useful in molecular biology
recognizes specific DNA sequences, and then cleave the DNA sequences somewhere else
cuts DNA at specific sequences to create sticky ends
“restrict” the entry of foreign DNA into the bacterial cells
Type II Restriction Endonucleases
restriction enzymes
cleave DNA within the recognition site
this property has made them incredibly useful in molecular biology
DNA sequences cut by them can be rejoined with ligases
ends are either 5’, 3’ overhang, or blunt

Difference Between Type I and II RE
Type I cuts far from its recognition site (hundreds of bases away) and is a large, multi-subunit enzyme with both cutting and methylating functions, requiring ATP and SAM,
Type II cuts within or very near the recognition site, is simpler, usually just a cutting enzyme, and is the most used in molecular biology for predictable results
Palindromic Sequence
seen in type II REs
sequence of nucleotide bases reads the same on the top strand as the sequence of nucleotide bases reads on the bottom strand of the DNA molecule in the 5’ → 3’ direction
RE Enzymes
EcoRI
BamHI
HindIII
EcoRI
Escherichi coli
strain RY13
1st endonuclease isolated
BamHI
Bacillus amyloliquefaciens
strain H
1st endonuclease
HindIII
Haemophilus influenzae
strain Rd
3rd endonuclease isolated
Why dont bacterial restriction endonucleases attack the host’s own DNA?
the most common reason is that the host bacterial cell methylates a base in every copy of the RE site within its own genome
Gel Electrophoresis
a method for sorting DNA and RNA sequence fragments by size
at neutral pH, DNA molecules are - charged because of phosphate groups
in an electrical field, DNA will tend to move towards the positive electrode
cannot be done in a liquid, as it needs to make a gel
most common kind is made from uncharged polysaccharide agrose
gel contains a buffer that provides ions a current which they can flow, and to keep the pH slightly above neutral
“printed” gel is stained with a DNA binding fluorescent dye, EtBr
Size Fractionation of DNA
shorter DNA fragments migrate more rapidly through the gel-matrix than longer molecules
migration rate of linear DNA molecule is inversely related to log of its molecular mass, or # of its base pairs
meaning, larger molecules travel less distance and visa versa
a standard curve of known size DNA fragments can be used to extrapolate the size of an unknown DNA fragment
often in the 1st stage in the characterization of an unknown DNA molecule
Factors That Affect Mobility of DNA Fragments in a Gel
molecular mass (bp) of a DNA molecule
agarose concentration in gel
topology of DNA molecule
voltage
Concentration of Agarose in a DNA Molecule
as agarose concentration increases, pore size in gel matrix decreases
smaller pores means more resistance to DNA movement, favouring small DNA fragments, and giving better resolution of size differences of small fragments
DNA Molecule Topologies
DNA molecules can exist in different topologies:
linear
relaxed
travels the least
circular
travels the least
supercoiled
travels the furthest
topology of DNA strand affects its rate of migration in gels
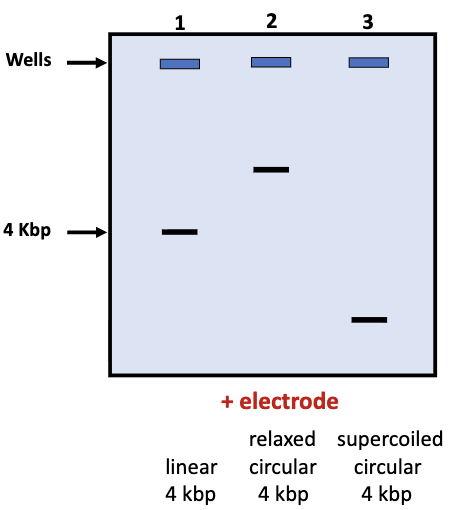
Supercoiled DNA
can either be circular or linear, but the ends of the linear molecule must be restrained
in cells, DNA is often negatively supercoiled
+ supercoiled DNA can be produced in vitro
Gel Electrophoresis Voltage
greater voltage speeds up migration rate of DNA fragments during agarose gel-electrophoresis
What is meant by ‘sticky ends’ produced by REs and how do they help when DNA fragments are rejoined using ligase to produce recombinant DNA
Minimum Requirements for DNA Synthesis in Vivo
a strand of DNA to act a a template
a short, single strand of DNA complementary to part of the template
DNA polymerase
dNTPs
Mg+
PCR
polymerase chain reaction
has been called the most important technique in molecular bio
enzymatic copying of double-stranded DNA using 2 primers, complementary to opposite strands could lead to exponential increase in amount of target sequence
requires DNA to be cycled repeatedly through 3 temperatures
this allows (assuming reaction occurs with 100% efficiency) for more than a billion-fold amplification of target DNA
Denaturation
temperature 94-96o
double stranded DNA denatures to single stranded DNA
Annealing
temperatures 50-65o C
primers bind to their complementary sequences
Tm is dependent on length and base composition of primers
Elongation/extension
temperature 72o C
DNA polymerase binds to the annealed primers and extends DNA at the 3’ end of the chain
PCR Ingredients
deoxyribonucleotide triphosphates (dNTP’s)
Mg2+
primers
template DNA
thermostable DNA polymerase (often Taq)
a salt
Tris (pH control)
stabilizers
PCR Primers
short molecule of single stranded DNA, most often 18-25 bp long
priming between two oligos (single stranded DNA) annealed to opposite strands can give exponential growth of product
size of PCR product depends on how far apart the annealing sites of the 2 primers are
Length of Primers
successful PCR primers are usually 18-25 bp long, as PCR depends on specific binding of primers to the exact positions that will allow us to amplify our target DNA
specificity of primer binding is related to primer length
shorter primers may not be specific enough in their binding, they may match and bind to multiple positions in the genomic DNA, resulting in amplification of incorrect DNA sequences
primers that are 18-25 bp long are long enough to match only the intended DNA target sequence
Applications of PCR
amplifying target sequences for further study
detection of rare DNA sequences
but not good for determining abundance of these rare sequences
Stages of PCR
during early cycles of PCR, production of DNA product is only limited by the amount in the previous cycle
exponential phase
in later cycles, dNTPs are less abundant, and our DNA polymerase may start to wear out, leading to slower growth of product
linear growth
eventually, growth in amount of PCR product slows down greatly and then stops, as polymerase and dNTPs start to become exhausted
plateau phase
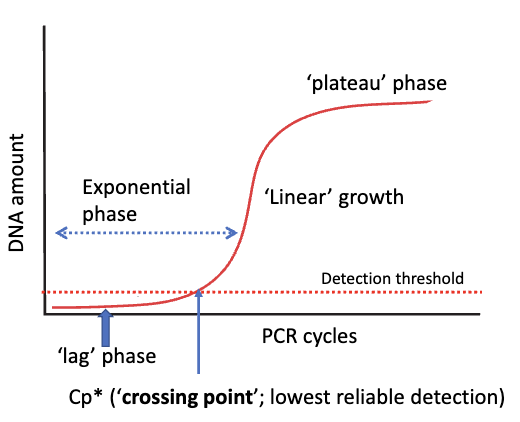
Cp value
marks the first point product exceeds detection threshold of instrument in PCR
qPCR
quantitative PCR
how DNA is quantified in each cycle
growth in amount of PCR product is monitored by using a reporter dye, and a PCR machine capable of detecting fluorescence in each well
amount of PCR product (in exponential phase of qPCR) is proportional to starting amount of DNA
SYBR Green
simplest and cheapest reporter dye in qPCR
SYBR fluoresces much more strongly when bound to double-stranded DNA
binds primarily to minor groove in double stranded DNA
Applications of qPCR
quantify amount of starting DNA of a particular sequence
measuring rate at which a particular gene is transcribed
PCR Set Up

What are the 5 minimum requirements for DNA synthesis in vitro?
DNA template
Primer (short ssDNA with free 3′-OH)
DNA polymerase
dNTPs (dATP, dCTP, dGTP, dTTP)
Mg²⁺ (essential cofactor for polymerase)
What are the three stages of a PCR cycle?
Denaturation (94–96 °C) – DNA strands separate
Annealing (50–65 °C) – primers bind
Extension / Elongation (72 °C) – polymerase synthesizes DNA
What happens to the target DNA content after each PCR cycle?
The amount of target DNA doubles (during exponential phase)
How many PCR cycles do you need? How many DNA copies are there?
need 30-35 cycles
Ideally: 2ⁿ, where n = number of cycles
(e.g., 30 cycles ≈ 1 billion copies, assuming perfect efficiency)
How do you “measure” the final amount of DNA product?
Regular PCR: end-point measurement (e.g., gel electrophoresis)
qPCR: fluorescence measured during each cycle
What is Taq
A thermostable DNA polymerase from Thermus aquaticus
Survives repeated heating to ~95 °C without denaturing
What direction is the new DNA strand synthesized (3’→5’ OR 5’→3’)?
5’ → 3’
In a mixture of DNA ... what provides the specificity of the target
sequence that will be amplified?
Primers
They determine:
Which DNA region is amplified
Product size
Specificity of binding
What is the “usual” size of DNA fragment that can be amplified by PCR?
Most commonly ≤ 2 kb
Can go up to ~40 kb, but efficiency drops
What is the “usual” primer length? Why not shorter/longer?
18-25 base pairs
Too short: bind nonspecifically → wrong products
Too long: expensive, little gain in specificity
An invasive species produces a novel toxic protein ... you have isolated
this protein using chromatography, but you still don’t know what it is.
You successfully isolate DNA from the invasive species ... can you amplify the gene for this toxic protein?
No — not without DNA sequence information
You must know enough of the DNA sequence to design primers
Is “regular” PCR good for determining the abundance of a particular DNA sequence in sample?
No
Because:
End-point PCR reaches a plateau
Final product ≠ starting amount
Draw a PCR amplification curve
Axes:
x-axis: PCR cycles
y-axis: DNA amount (or fluorescence)
Four phases:
Lag phase
Exponential phase
Linear phase
Plateau phase
What is the relationship between the “exponential” phase and the “log-linear” phase?
They are the same phase, just plotted differently
What are the differences between the “linear” phase, and the “log-linear”
phase?
Log-linear: high precision, constant doubling
Linear: reagents limiting, variable growth
Which phase allows us to quantify the starting amount of DNA?
Exponential phase (log-linear)
snapshots via fluorescent dye
able to use a machine, as Cp is not yet reached
How do we monitor the growth of the product from one cycle to another?
Fluorescent reporter dyes
SYBR Green (binds dsDNA)
Fluorescent probes (target-specific)
What are the two key features that distinguish qPCR from (regular) PCR?
Measures DNA during each cycle
Allows quantification of starting DNA
Sanger Dideoxy Chain Terminating Method
one of the two methods of sequencing DNA
invented in the 1970s, and still used today
remains the cold standard for accuracy and conviencience for sequencing small numbers of samples
Dideoxyribonucleoside Triphosphates
ddNTP
terminate DNA synthesis
whereas normal dNTP extends the DNA strand
consider a DNA synthesis reaciton where 5% of the dGTP is replaced with ddGTP. this would give us DNA daughter strands of varying lengths, the lengths of which are determined by where the G’s occur in the sequence
we could do the same thing for the other bases (ddATP, CTP, TTP) so that we would get a subset of DNA elongation products terminating with a ddNTP base at every position in the DNA sequence
how would we keep track of which bases are terminating which fragments?
how do we sort out the different fragments by size?
How do we keep track of which bases are terminated by which fragments (ddNTP)?
Fluorescent dideoxy sequencing
we attach different fluorescent colours to each type of ddNTP, then use gel electrophoresis to sort the fragments by size.
the smallest fragments represent DNA sequences terminating close to the primer
as ddNTP-terminated fragments migrate in the gel, they pass a laser beam, that excites the fluorescent dyes and a camera records the flash of coloured light that results
Sanger Dideozy Sequencing PROS
very accurate
relatively long sequencing reads
easy to do, and can be automatefd
low cost
Sanger Dideozy Sequencing CONS
too slow for may applications
ex. genome sequencing
costly when scaled up to aqquire lots of data
requires purification and preparation of each individual DNA sequence that is being studied
Human Genome Project
first human genome sequencing cost nearly $3 billion
mainly because the Sanger dideoxy sequencing was too slow and costly
now a human genome sequence costs less than $1000
this is due to the switch to massively parallel sequencing
Massively Parallel Sequencing
genomes are big!
handling and sequencing individual samples is too slow for genome sequencing
an approach was needed that allowed for many of DNA segments to be sequenced at once, and sequencing by synthesis
illumina
ion torrent
Illumina DNA Sequencing
DNA needs to be short segments
this is accomplished by shearing/use of short PCR productrs
adaptor sequences are added
DNA segments are sequenced to be randomly arrayed across the flow cell surface
bridge amplification is used to amplify a single DNA molecule into clusters of identical DNA molecules
sequencing occurs by the addition of fluorescently labeled nucleotide analogs, 1 base at a time.
these dNTP analogs are chain terminators (like Sanger) but are reversible (unlike Sanger)
after chemical treatment of the newly added dNTP, the chain can continue to elongate
after each dNTP is added, sequencer pauses and exposes flow cell to a laser, and takes a picture to record what base was incorporated in each cluster.
process continues for a few hundred cycles
computer interprets the data to infer the DNA sequence within each DNA cluster on the flow cell
millions of distinct DNA sequences determined simultaneously this way
massively parallel DNA sequencing
Illumina Adaptor Sequences
added by ligation to the ends of DNA segments
adaptors add sites for attachment of DNA sequencing primers, and enable attachment to the oligionucleotides on the surface of the flow cell
3rd Generation Sequencing
faster, single molecule, longer reads
Nanopore sequencing
Nanopore Sequencing
not DNA sequencing by synthesis
single molecule at a time, therefore no pre-amplification by PCR
enzyme unwinds DNA, a single strand is pulled by an electrical current through a pore in a membrane
each base produces a characteristic disturbance in electrical current, which can be used to read the base as it travels through the pore.
Nanopore PROS
long reads up to 1000 kb
no amplification step to boost amount of template DNA before sequencing
small, highly portable DNA sequencer connects to USB port on a computer
can be used in the field to get rapid results
can detect methylated bases
Nanopore CONS
slightly less accurate than other methods
Is Each Method Massively Parallel?
Sanger
No
Illumina
Yes
Nanopore
Yes
Is Each Method Sequencing by Synthesis?
Sanger
Yes
Illumina
Yes
Nanopore
No
Is Each Method Single Molecule
Sanger
No
Illumina
No
Nanopore
Yes
Is Each Method Chain Terminator?
Sanger
yes
non reversible
Illumina
yes
reversible
Nanopore
no
Is Each Method Accurate?
Sanger
most accurate
Illumina
middle
Nanopore
least accurate
still up to 98-99% accurate
Read Length of Each Method
Sanger
650-1000 bp
Illumina
75-600 bp
Nanopore
100 kb